
Entrez 3630 | Ensembl ENSG00000254647 | |
 | ||
Aliases INS, IDDM, IDDM1, IDDM2, ILPR, IRDN, MODY10, insulin External IDs MGI: 96573 HomoloGene: 173 GeneCards: INS | ||
Insulin (from the Latin, insula meaning island) is a peptide hormone produced by beta cells of the pancreatic islets. It regulates the metabolism of carbohydrates, fats and protein by promoting the absorption of, especially, glucose from the blood into fat, liver and skeletal muscle cells. In these tissues the absorbed glucose is converted into either glycogen via glycogenesis or fats (triglycerides) via lipogenesis, or, in the case of the liver, into both. Glucose production (and excretion into the blood) by the liver is strongly inhibited by high concentrations of insulin in the blood. Circulating insulin also affects the synthesis of proteins in a wide variety of tissues. It is therefore an anabolic hormone, promoting the conversion of small molecules in the blood into large molecules inside the cells. Low insulin levels in the blood have the opposite effect by promoting widespread catabolism.
Contents
- Evolution and species distribution
- Gene
- Alleles
- Regulation
- Protein structure
- Synthesis
- Release
- Oscillations
- Blood content
- Signal transduction
- Physiological effects
- Degradation
- Regulator of endocannabinoid metabolism
- Hypoglycemia
- Diseases and syndromes
- Medical uses
- Discovery
- Extraction and purification
- Nobel Prizes
- Nobel Prize controversy
- References
Pancreatic beta cells (β cells) are known to be sensitive to glucose concentrations in the blood. When glucose concentrations in the blood are high, the pancreatic β cells secrete insulin into the blood; when glucose levels are low, secretion of insulin is inhibited. Their neighboring alpha cells, by taking their cues from the beta cells, secrete glucagon into the blood in the opposite manner: increased secretion when blood glucose is low, and decreased secretion when glucose concentrations are high. Glucagon, through stimulating the liver to release glucose by glycogenolysis and gluconeogenesis, has the opposite effect of insulin. The secretion of insulin and glucagon into the blood in response to the blood glucose concentration is the primary mechanism responsible for keeping the glucose levels in the extracellular fluids within very narrow limits at rest, after meals, and during exercise and starvation.
If pancreatic beta cells are destroyed by an autoimmune reaction, insulin can no longer be synthesized or be secreted into the blood. This results in type 1 diabetes mellitus, which is characterized by abnormally high blood glucose concentrations, and generalized body wasting. In type 2 diabetes mellitus the destruction of beta cells is less pronounced than in type 1 diabetes, and is not due to an autoimmune process. Instead there is an accumulation of amyloid in the pancreatic islets, which disrupts their anatomy and physiology. Type 2 diabetes is characterized by high rates of glucagon secretion into the blood which are unaffected by, and unresponsive to the concentration of glucose in the blood glucose. Insulin is still secreted into the blood in response to the blood glucose. As a result, the insulin levels, even when the blood sugar level is normal, are much higher than they are in healthy persons. There are a variety of treatment regimens, none of which is entirely satisfactory. When the pancreas’s capacity to secrete insulin can no longer keep the blood sugar level within normal bounds, insulin injections are given.
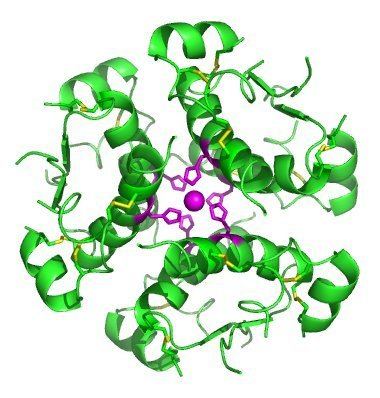
The human insulin protein is composed of 51 amino acids, and has a molecular mass of 5808 Da. It is a dimer of an A-chain and a B-chain, which are linked together by disulfide bonds. Insulin's structure varies slightly between species of animals. Insulin from animal sources differs somewhat in effectiveness (in carbohydrate metabolism effects) from human insulin because of these variations. Porcine insulin is especially close to the human version, and was widely used to treat type 1 diabetics before human insulin could be produced in large quantities by recombinant DNA technologies.
The crystal structure of insulin in the solid state was determined by Dorothy Hodgkin. It is on the WHO Model List of Essential Medicines, the most important medications needed in a basic health system.
Evolution and species distribution
Insulin may have originated more than a billion years ago. The molecular origins of insulin go at least as far back as the simplest unicellular eukaryotes. Apart from animals, insulin-like proteins are also known to exist in the Fungi and Protista kingdoms.
Insulin is produced by beta cells of the pancreatic islets in most vertebrates and by the Brockmann body in some teleost fish. Cone snails Conus geographus and Conus tulipa, venomous sea snails that hunt small fish, use modified forms of insulin in their venom cocktails. The insulin toxin, closer in structure to fishes' than to snails' native insulin, slows down the prey fishes by lowering their blood glucose levels.
Gene
The preproinsulin precursor of insulin is encoded by the INS gene.
Alleles
A variety of mutant alleles with changes in the coding region have been identified. A read-through gene, INS-IGF2, overlaps with this gene at the 5' region and with the IGF2 gene at the 3' region.
Regulation
Several regulatory sequences in the promoter region of the human insulin gene bind to transcription factors. In general, the A-boxes bind to Pdx1 factors, E-boxes bind to NeuroD, C-boxes bind to MafA, and cAMP response elements to CREB. There are also silencers that inhibit transcription.
Protein structure
Within vertebrates, the amino acid sequence of insulin is strongly conserved. Bovine insulin differs from human in only three amino acid residues, and porcine insulin in one. Even insulin from some species of fish is similar enough to human to be clinically effective in humans. Insulin in some invertebrates is quite similar in sequence to human insulin, and has similar physiological effects. The strong homology seen in the insulin sequence of diverse species suggests that it has been conserved across much of animal evolutionary history. The C-peptide of proinsulin (discussed later), however, differs much more among species; it is also a hormone, but a secondary one.
The primary structure of bovine insulin was first determined by Frederick Sanger in 1951. After that, this polypeptide was synthesized independently by several groups. The 3-dimensional structure of insulin was determined by X-ray crystallography in Dorothy Hodgkin's laboratory in 1969 (PDB file 1ins).
Insulin is produced and stored in the body as a hexamer (a unit of six insulin molecules), while the active form is the monomer. The hexamer is an inactive form with long-term stability, which serves as a way to keep the highly reactive insulin protected, yet readily available. The hexamer-monomer conversion is one of the central aspects of insulin formulations for injection. The hexamer is far more stable than the monomer, which is desirable for practical reasons; however, the monomer is a much faster-reacting drug because diffusion rate is inversely related to particle size. A fast-reacting drug means insulin injections do not have to precede mealtimes by hours, which in turn gives people with diabetes more flexibility in their daily schedules. Insulin can aggregate and form fibrillar interdigitated beta-sheets. This can cause injection amyloidosis, and prevents the storage of insulin for long periods.
Synthesis
Insulin is produced in the pancreas and Brockmann body, and released when any of several stimuli are detected. These stimuli include ingested protein and glucose in the blood produced from digested food. Carbohydrates can be polymers of simple sugars or the simple sugars themselves. If the carbohydrates include glucose, then that glucose will be absorbed into the bloodstream and blood glucose level will begin to rise. In target cells, insulin initiates a signal transduction, which has the effect of increasing glucose uptake and storage. Finally, insulin is degraded, terminating the response.
In mammals, insulin is synthesized in the pancreas within the β-cells of the islets of Langerhans. One million to three million islets of Langerhans (pancreatic islets) form the endocrine part of the pancreas, which is primarily an exocrine gland. The endocrine portion accounts for only 2% of the total mass of the pancreas. Within the islets of Langerhans, beta cells constitute 65–80% of all the cells.
Insulin consists of two polypeptide chains, the A- and B- chains, linked together by disulfide bonds. It is however first synthesized as a single polypeptide called preproinsulin in pancreatic β-cells. Preproinsulin contains a 24-residue signal peptide which directs the nascent polypeptide chain to the rough endoplasmic reticulum (RER). The signal peptide is cleaved as the polypeptide is translocated into lumen of the RER, forming proinsulin. In the RER the proinsulin folds into the correct conformation and 3 disulfide bonds are formed. About 5–10 min after its assembly in the endoplasmic reticulum, proinsulin is transported to the trans-Golgi network (TGN) where immature granules are formed. Transport to the TGN may take about 30 min.
Proinsulin undergoes maturation into active insulin through the action of cellular endopeptidases known as prohormone convertases (PC1 and PC2), as well as the exoprotease carboxypeptidase E. The endopeptidases cleave at 2 positions, releasing a fragment called the C-peptide, and leaving 2 peptide chains, the B- and A- chains, linked by 2 disulfide bonds. The cleavage sites are each located after a pair of basic residues (lysine-64 and arginine-65, and arginine-31 and -32). After cleavage of the C-peptide, these 2 pairs of basic residues are removed by the carboxypeptidase. The C-peptide is the central portion of proinsulin, and the primary sequence of proinsulin goes in the order "B-C-A" (the B and A chains were identified on the basis of mass and the C-peptide was discovered later).
The resulting mature insulin is packaged inside mature granules waiting for metabolic signals (such as leucine, arginine, glucose and mannose) and vagal nerve stimulation to be exocytosed from the cell into the circulation.
The endogenous production of insulin is regulated in several steps along the synthesis pathway:
Insulin and its related proteins have been shown to be produced inside the brain, and reduced levels of these proteins are linked to Alzheimer's disease.
Release
Beta cells in the islets of Langerhans release insulin in two phases. The first-phase release is rapidly triggered in response to increased blood glucose levels, and lasts about 10 minutes. The second phase is a sustained, slow release of newly formed vesicles triggered independently of sugar, peaking in 2 to 3 hours. Reduced first-phase insulin release may be the earliest detectable beta cell defect predicting onset of type 2 diabetes. First-phase release and insulin sensitivity are independent predictors of diabetes.
The description of first phase release is as follows:
This is the primary mechanism for release of insulin. Other substances known to stimulate insulin release include the amino acids arginine and leucine, parasympathetic release of acetylcholine (acting via the phospholipase C pathway), sulfonylurea, cholecystokinin (CCK, also via phospholipase C), and the gastrointestinally derived incretins, such as glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic peptide (GIP).
Release of insulin is strongly inhibited by norepinephrine (noradrenaline), which leads to increased blood glucose levels during stress. It appears that release of catecholamines by the sympathetic nervous system has conflicting influences on insulin release by beta cells, because insulin release is inhibited by α2-adrenergic receptors and stimulated by β2-adrenergic receptors. The net effect of norepinephrine from sympathetic nerves and epinephrine from adrenal glands on insulin release is inhibition due to dominance of the α-adrenergic receptors.
When the glucose level comes down to the usual physiologic value, insulin release from the β-cells slows or stops. If the blood glucose level drops lower than this, especially to dangerously low levels, release of hyperglycemic hormones (most prominently glucagon from islet of Langerhans alpha cells) forces release of glucose into the blood from the liver glycogen stores, supplemented by gluconeogenesis if the glycogen stores become depleted. By increasing blood glucose, the hyperglycemic hormones prevent or correct life-threatening hypoglycemia.
Evidence of impaired first-phase insulin release can be seen in the glucose tolerance test, demonstrated by a substantially elevated blood glucose level at 30 minutes after the ingestion of a glucose load (75 or 100 g of glucose), followed a slow drop over the next 100 minutes, to remain above 120 mg/100 ml after two hours after the start of the test. In a normal person the blood glucose level is corrected (and may even be slightly over-corrected) by the end of the test.
Oscillations
Even during the digestion, in general, one or two hours following a meal, insulin release from the pancreas is not continuous, but oscillates with a period of 3–6 minutes, changing from generating a blood insulin concentration more than about 800 pmol/l to less than 100 pmol/l. This is thought to avoid downregulation of insulin receptors in target cells, and to assist the liver in extracting insulin from the blood. This oscillation is important to consider when administering insulin-stimulating medication, since it is the oscillating blood concentration of insulin release, which should, ideally, be achieved, not a constant high concentration. This may be achieved by delivering insulin rhythmically to the portal vein or by islet cell transplantation to the liver. It is hoped that future insulin pumps will address this characteristic. (See also Pulsatile Insulin.)
Blood content
The blood content of insulin can be measured in international units, such as µIU/mL or in molar concentration, such as pmol/L, where 1 µIU/mL equals 6.945 pmol/L. A typical blood level between meals is 8–11 μIU/mL (57–79 pmol/L).
Signal transduction
The manner in which the presence of insulin in the extracellular fluids affects metabolic changes inside its target cells is strangely complex and counterintuitive. Insulin binds to the extracellular portion of cell membrane-bound insulin receptors. Before insulin binds to its receptor, however, the receptor consists of two identical protein monomers, which remain separate until insulin binding occurs. In the presence of insulin in the extracellular fluid, two of these monomers come together, bind the insulin molecule (also a protein) to their unified extracellular domains, resulting in the conversion of the intracellular domain of the resulting protein complex into an active enzyme, tyrosine kinase. This enzyme then phosphorylates itself, as well as a cytosolic protein named insulin receptor substrate 1 (IRS-1), which becomes bound to activated insulin receptor. The resulting complex sets a variety of intracellular phosphorylation cascades in motion which ultimately result in the insertion of glucose transporter type 4 (GLUT4) molecules into the cell membranes of fat and muscle tissues, increasing the rate of glucose transport into these cells (making their glucose uptake “insulin dependent”). It also results in the activation of the enzyme, glycogen synthase, causing liver and muscle cells to convert glucose into glycogen. A similar activation of the glycolytic enzymes, and of acetyl CoA carboxylase, stimulates liver, adipose and lactating mammary gland tissue to synthesize triglycerides (fats). Another phosphorylation cascade that is activated by insulin binding to its receptor is the multistep Erk and MAP kinase pathways, which ultimately phosphorylate a number of substrates important for cell proliferation, cell cycle progression, cell division and differentiation (RSK kinases, Elk-1 transcription factor, etc.)
The cascade that leads to the insertion of GLUT4 glucose transporters into the cell membranes of muscle and fat cells, and to the synthesis of glycogen in liver and muscle tissue, as well as the conversion of glucose into triglycerides in liver, adipose, and lactating mammary gland tissue, operates via the activation, by IRS-1, of phosphoinositol 3 kinase (PI3K). This enzyme converts a phospholipid in the cell membrane by the name of phosphatidylinositol 4,5-bisphosphate (PIP2), into phosphatidylinositol 3,4,5-triphosphate (PIP3), which, in turn, activates protein kinase B (PKB). Activated PKB facilitates the fusion of GLUT4 containing endosomes with the cell membrane, resulting in an increase in GLUT4 transporters in the plasma membrane. PKB also phosphorylates glycogen synthase kinase (GSK), thereby inactivating this enzyme. This means that its substrate, glycogen synthase (GS), cannot be phosphorylated, and remains dephosphorylated, and therefore active. The active enzyme, glycogen synthase (GS), catalyzes the rate limiting step in the synthesis of glycogen from glucose. Similar dephosphorylations affect the enzymes controlling the rate of glycolysis leading to the synthesis of fats via malonyl-CoA in the tissues that can generate triglycerides, and also the enzymes that control the rate of gluconeogenesis in the liver. The overall effect of these final enzyme dephosphorylations is that, in the tissues that can carry out these reactions, glycogen and fat synthesis from glucose are stimulated, and glucose production by the liver through glycogenolysis and gluconeogenesis are inhibited. The breakdown of triglycerides by adipose tissue into free fatty acids and glycerol is also inhibited.
After the intracellular signal that resulted from the binding of insulin to its receptor has been produced, termination of signaling is then needed. As mentioned below in the section on degradation, endocytosis and degradation of the receptor bound to insulin is a main mechanism to end signaling. In addition, the signaling pathway is also terminated by dephosphorylation of the tyrosine residues in the various signaling pathways by tyrosine phosphatases. Serine/Threonine kinases are also known to reduce the activity of insulin.
The structure of the insulin–insulin receptor complex has been determined using the techniques of X-ray crystallography.
Physiological effects
The actions of insulin on the global human metabolism level include:
The actions of insulin (indirect and direct) on cells include:
Insulin also influences other body functions, such as vascular compliance and cognition. Once insulin enters the human brain, it enhances learning and memory and benefits verbal memory in particular. Enhancing brain insulin signaling by means of intranasal insulin administration also enhances the acute thermoregulatory and glucoregulatory response to food intake, suggesting that central nervous insulin contributes to the co-ordination of a wide variety of homeostatic or regulatory processes in the human body. Insulin also has stimulatory effects on gonadotropin-releasing hormone from the hypothalamus, thus favoring fertility.
Degradation
Once an insulin molecule has docked onto the receptor and effected its action, it may be released back into the extracellular environment, or it may be degraded by the cell. The two primary sites for insulin clearance are the liver and the kidney. The liver clears most insulin during first-pass transit, whereas the kidney clears most of the insulin in systemic circulation. Degradation normally involves endocytosis of the insulin-receptor complex, followed by the action of insulin-degrading enzyme. An insulin molecule produced endogenously by the pancreatic beta cells is estimated to be degraded within about one hour after its initial release into circulation (insulin half-life ~ 4–6 minutes).
Regulator of endocannabinoid metabolism
Insulin is a major regulator of endocannabinoid (EC) metabolism and insulin treatment have shown to reduce intracellular ECs, the 2-arachidonylglycerol (2-AG) and anandamide (AEA), which correspond with insulin-sensitive expression changes in enzymes of EC metabolism. In insulin-resistant adipocytes, patterns of insulin-induced enzyme expression is disturbed in a manner consistent with elevated EC synthesis and reduced EC degradation. Findings suggest that insulin-resistant adipocytes fail to regulate EC metabolism and decrease intracellular EC levels in response to insulin stimulation, whereby obese insulin-resistant individuals exhibit increased concentrations of ECs. This dysregulation contribute to excessive visceral fat accumulation and reduced adiponectin release from abdominal adipose tissue, and further to the onset of several cardiometabolic risk factors that are associated with obesity and type 2 diabetes.
Hypoglycemia
Although other cells can use other fuels (most prominently fatty acids), neurons depend on glucose as a source of energy, unless the person is in ketosis. They do not require insulin to absorb glucose, unlike muscle and adipose tissue, and they have very small internal stores of glycogen. Glycogen stored in liver cells (unlike glycogen stored in muscle cells) can be converted to glucose, and released into the blood, when glucose from digestion is low or absent, and the glycerol backbone in triglycerides can also be used to produce blood glucose.
Sufficient lack of glucose and scarcity of these sources of glucose can dramatically make itself manifest in the impaired functioning of the central nervous system: dizziness, speech problems, and even loss of consciousness. Low blood glucose level is known as hypoglycemia or, in cases producing unconsciousness, "hypoglycemic coma" (sometimes termed "insulin shock" from the most common causative agent). Endogenous causes of insulin excess (such as an insulinoma) are very rare, and the overwhelming majority of insulin excess-induced hypoglycemia cases are iatrogenic and usually accidental. A few cases of murder, attempted murder, or suicide using insulin overdoses have been reported, but most insulin shocks appear to be due to errors in dosage of insulin (e.g., 20 units instead of 2) or other unanticipated factors (did not eat as much as anticipated, or exercised more than expected, or unpredicted kinetics of the subcutaneously injected insulin itself).
Possible causes of hypoglycemia include:
Diseases and syndromes
There are several conditions in which insulin disturbance is pathologic:
Medical uses
Biosynthetic human insulin (insulin human rDNA, INN) for clinical use is manufactured by recombinant DNA technology. Biosynthetic human insulin has increased purity when compared with extractive animal insulin, enhanced purity reducing antibody formation. Researchers have succeeded in introducing the gene for human insulin into plants as another method of producing insulin ("biopharming") in safflower. This technique is anticipated to reduce production costs.
Several analogs of human insulin are available. These insulin analogs are closely related to the human insulin structure, and were developed for specific aspects of glycemic control in terms of fast action (prandial insulins) and long action (basal insulins). The first biosynthetic insulin analog was developed for clinical use at mealtime (prandial insulin), Humalog (insulin lispro), it is more rapidly absorbed after subcutaneous injection than regular insulin, with an effect 15 minutes after injection. Other rapid-acting analogues are NovoRapid and Apidra, with similar profiles. All are rapidly absorbed due to sequence that will reduce formation of dimers and hexamers (monomeric insulins are more rapidly absorbed). Fast acting insulins do not require the injection-to-meal interval previously recommended for human insulin and animal insulins. The other type is long acting insulin; the first of these was Lantus (insulin glargine). These have a steady effect for an extended period from 18 to 24 hours. Likewise, another protracted insulin analogue (Levemir) is based on a fatty acid acylation approach. A myristyric acid molecule is attached to this analogue, which in turn associates the insulin molecule to the abundant serum albumin, which in turn extends the effect and reduces the risk of hypoglycemia. Both protracted analogues need to be taken only once daily, and are used for type 1 diabetics as the basal insulin. A combination of a rapid acting and a protracted insulin is also available, making it more likely for patients to achieve an insulin profile that mimics that of the body´s own insulin release.
Insulin is usually taken as subcutaneous injections by single-use syringes with needles, via an insulin pump, or by repeated-use insulin pens with disposable needles. Inhaled insulin is also available in U.S. market now.
Synthetic insulin can trigger adverse effects and so some people with diabetes rely on animal-source inulin.
Unlike many medicines, insulin currently cannot be taken orally because, like nearly all other proteins introduced into the gastrointestinal tract, it is reduced to fragments, whereupon all activity is lost. There has been some research into ways to protect insulin from the digestive tract, so that it can be administered orally or sublingually.
Discovery
In 1869, while studying the structure of the pancreas under a microscope, Paul Langerhans, a medical student in Berlin, identified some previously unnoticed tissue clumps scattered throughout the bulk of the pancreas. The function of the "little heaps of cells", later known as the islets of Langerhans, initially remained unknown, but Edouard Laguesse later suggested they might produce secretions that play a regulatory role in digestion. Paul Langerhans' son, Archibald, also helped to understand this regulatory role. The term "insulin" originates from insula, the Latin word for islet/island.
In 1889, the Polish-German physician Oskar Minkowski, in collaboration with Joseph von Mering, removed the pancreas from a healthy dog to test its assumed role in digestion. Several days after the removal of the dog's pancreas, Minkowski's animal-keeper noticed a swarm of flies feeding on the dog's urine. On testing the urine, they found sugar, establishing for the first time a relationship between the pancreas and diabetes. In 1901 Eugene Lindsay Opie took another major step forward when he clearly established the link between the islets of Langerhans and diabetes: "Diabetes mellitus ... is caused by destruction of the islets of Langerhans and occurs only when these bodies are in part or wholly destroyed". Before Opie's work, medical science had clearly established the link between the pancreas and diabetes, but not the specific role of the islets.
Over the next two decades researchers made several attempts to isolate – as a potential treatment – whatever the islets produced. In 1906 George Ludwig Zuelzer achieved partial success in treating dogs with pancreatic extract, but he was unable to continue his work. Between 1911 and 1912, E.L. Scott at the University of Chicago used aqueous pancreatic extracts, and noted "a slight diminution of glycosuria", but was unable to convince his director of his work's value; it was shut down. Israel Kleiner demonstrated similar effects at Rockefeller University in 1915, but World War I interrupted his work and he did not return to it.
In 1916, Nicolae Paulescu, a Romanian professor of physiology at the University of Medicine and Pharmacy in Bucharest, developed an aqueous pancreatic extract which, when injected into a diabetic dog, had a normalizing effect on blood-sugar levels. He had to interrupt his experiments because of World War I, and in 1921 he wrote four papers about his work carried out in Bucharest and his tests on a diabetic dog. Later that year, he published "Research on the Role of the Pancreas in Food Assimilation".
Extraction and purification
In October 1920, Canadian Frederick Banting concluded that it was the very digestive secretions that Minkowski had originally studied that were breaking down the islet secretion(s), thereby making it impossible to extract successfully. He jotted a note to himself: "Ligate pancreatic ducts of the dog. Keep dogs alive till acini degenerate leaving islets. Try to isolate internal secretion of these and relieve glycosurea."
The idea was the pancreas's internal secretion, which, it was supposed, regulates sugar in the bloodstream, might hold the key to the treatment of diabetes. A surgeon by training, Banting knew certain arteries could be tied off that would lead to atrophy of most of the pancreas, while leaving the islets of Langerhans intact. He theorized a relatively pure extract could be made from the islets once most of the rest of the pancreas was gone.
In the spring of 1921, Banting traveled to Toronto to explain his idea to J.J.R. Macleod, who was Professor of Physiology at the University of Toronto, and asked Macleod if he could use his lab space to test the idea. Macleod was initially skeptical, but eventually agreed to let Banting use his lab space while he was on holiday for the summer. He also supplied Banting with ten dogs on which to experiment, and two medical students, Charles Best and Clark Noble, to use as lab assistants, before leaving for Scotland. Since Banting required only one lab assistant, Best and Noble flipped a coin to see which would assist Banting for the first half of the summer. Best won the coin toss, and took the first shift as Banting's assistant. Loss of the coin toss may have proved unfortunate for Noble, given that Banting decided to keep Best for the entire summer, and eventually shared half his Nobel Prize money and a large part of the credit for the discovery of insulin with the winner of the toss. Had Noble won the toss, his career might have taken a different path. Banting's method was to tie a ligature around the pancreatic duct; when examined several weeks later, the pancreatic digestive cells had died and been absorbed by the immune system, leaving thousands of islets. They then isolated an extract from these islets, producing what they called "isletin" (what we now know as insulin), and tested this extract on the dogs starting July 27. Banting and Best were then able to keep a pancreatectomized dog named Marjorie alive for the rest of the summer by injecting her with the crude extract they had prepared. Removal of the pancreas in test animals in essence mimics diabetes, leading to elevated blood glucose levels. Marjorie was able to remain alive because the extracts, containing isletin, were able to lower her blood glucose levels.
Banting and Best presented their results to Macleod on his return to Toronto in the fall of 1921, but Macleod pointed out flaws with the experimental design, and suggested the experiments be repeated with more dogs and better equipment. He then supplied Banting and Best with a better laboratory, and began paying Banting a salary from his research grants. Several weeks later, the second round of experiments was also a success; and Macleod helped publish their results privately in Toronto that November. However, they needed six weeks to extract the isletin, which forced considerable delays. Banting suggested they try to use fetal calf pancreas, which had not yet developed digestive glands; he was relieved to find this method worked well. With the supply problem solved, the next major effort was to purify the extract. In December 1921, Macleod invited the biochemist James Collip to help with this task, and, within a month, the team felt ready for a clinical test.
On January 11, 1922, Leonard Thompson, a 14-year-old diabetic who lay dying at the Toronto General Hospital, was given the first injection of insulin. However, the extract was so impure, Thompson suffered a severe allergic reaction, and further injections were canceled. Over the next 12 days, Collip worked day and night to improve the ox-pancreas extract, and a second dose was injected on January 23. This was completely successful, not only in having no obvious side-effects but also in completely eliminating the glycosuria sign of diabetes. The first American patient was Elizabeth Hughes Gossett, the daughter of U.S. Secretary of State Charles Evans Hughes. The first patient treated in the U.S. was future woodcut artist James D. Havens; Dr. John Ralston Williams imported insulin from Toronto to Rochester, New York, to treat Havens.
Children dying from diabetic ketoacidosis were kept in large wards, often with 50 or more patients in a ward, mostly comatose. Grieving family members were often in attendance, awaiting the (until then, inevitable) death.
Banting and Best never worked well with Collip, regarding him as something of an interloper, and Collip left the project soon after.
Over the spring of 1922, Best managed to improve his techniques to the point where large quantities of insulin could be extracted on demand, but the preparation remained impure. The drug firm Eli Lilly and Company had offered assistance not long after the first publications in 1921, and they took Lilly up on the offer in April. In November, Lilly made a major breakthrough and was able to produce large quantities of highly refined insulin. Insulin was offered for sale shortly thereafter.
Synthesis
Purified animal-sourced insulin was the only type of insulin available to diabetics until genetic advances occurred later with medical research. The amino acid structure of insulin was characterized in the early 1950s by Frederick Sanger, and the first synthetic insulin was produced simultaneously in the labs of Panayotis Katsoyannis at the University of Pittsburgh and Helmut Zahn at RWTH Aachen University in the early 1960s.
The first genetically engineered, synthetic "human" insulin was produced using E. coli in 1978 by Arthur Riggs and Keiichi Itakura at the Beckman Research Institute of the City of Hope in collaboration with Herbert Boyer at Genentech. Genentech, founded by Swanson, Boyer and Eli Lilly and Company, went on in 1982 to sell the first commercially available biosynthetic human insulin under the brand name Humulin. The vast majority of insulin currently used worldwide is now biosynthetic recombinant "human" insulin or its analogues.
Recombinant insulin is produced either in yeast (usually Saccharomyces cerevisiae) or E. coli. In yeast, insulin may be engineered as a single-chain protein with a KexII endoprotease (a yeast homolog of PCI/PCII) site that separates the insulin A chain from a c-terminally truncated insulin B chain. A chemically synthesized c-terminal tail is then grafted onto insulin by reverse proteolysis using the inexpensive protease trypsin; typically the lysine on the c-terminal tail is protected with a chemical protecting group to prevent proteolysis. The ease of modular synthesis and the relative safety of modifications in that region accounts for common insulin analogs with c-terminal modifications (e.g. lispro, aspart, glulisine). The Genentech synthesis and completely chemical synthesis such as that by Bruce Merrifield are not preferred because the efficiency of recombining the two insulin chains is low, primarily due to competition with the precipitation of insulin B chain.
Nobel Prizes
The Nobel Prize committee in 1923 credited the practical extraction of insulin to a team at the University of Toronto and awarded the Nobel Prize to two men: Frederick Banting and J.J.R. Macleod. They were awarded the Nobel Prize in Physiology or Medicine in 1923 for the discovery of insulin. Banting, insulted that Best was not mentioned, shared his prize with him, and Macleod immediately shared his with James Collip. The patent for insulin was sold to the University of Toronto for one half-dollar.
The primary structure of insulin was determined by British molecular biologist Frederick Sanger. It was the first protein to have its sequence be determined. He was awarded the 1958 Nobel Prize in Chemistry for this work.
In 1969, after decades of work, Dorothy Hodgkin determined the spatial conformation of the molecule, the so-called tertiary structure, by means of X-ray diffraction studies. She had been awarded a Nobel Prize in Chemistry in 1964 for the development of crystallography.
Rosalyn Sussman Yalow received the 1977 Nobel Prize in Medicine for the development of the radioimmunoassay for insulin.
George Minot, co-recipient of the 1934 Nobel Prize for the development of the first effective treatment for pernicious anemia, had diabetes mellitus. Dr. William Castle observed that the 1921 discovery of insulin, arriving in time to keep Minot alive, was therefore also responsible for the discovery of a cure for pernicious anemia.
Nobel Prize controversy
The work published by Banting, Best, Collip and Macleod represented the preparation of purified insulin extract suitable for use on human patients. Although Paulescu discovered the principles of the treatment, his saline extract could not be used on humans; he was not mentioned in the 1923 Nobel Prize. Professor Ian Murray was particularly active in working to correct "the historical wrong" against Nicolae Paulescu. Murray was a professor of physiology at the Anderson College of Medicine in Glasgow, Scotland, the head of the department of Metabolic Diseases at a leading Glasgow hospital, vice-president of the British Association of Diabetes, and a founding member of the International Diabetes Federation. Murray wrote:
Insufficient recognition has been given to Paulescu, the distinguished Romanian scientist, who at the time when the Toronto team were commencing their research had already succeeded in extracting the antidiabetic hormone of the pancreas and proving its efficacy in reducing the hyperglycaemia in diabetic dogs.
In a private communication Professor Tiselius, former head of the Nobel Institute, expressed his personal opinion that Paulescu was equally worthy of the award in 1923.