
 | ||
While the cellular and molecular mechanisms of learning and memory have long been a central focus of neuroscience, it is only in recent years that attention has turned to the epigenetic mechanisms behind the dynamic changes in gene transcription responsible for memory formation and maintenance. Epigenetic gene regulation often involves the physical marking (chemical modification) of DNA or associated proteins to cause or allow long-lasting changes in gene activity. Epigenetic mechanisms such as DNA methylation and histone modifications (methylation, acetylation, and deacetylation) have been shown to play an important role in learning and memory.
Contents
- DNA Methylation
- DNMTs and Memory
- DNA Methylation Targets
- Demethylation and Memory
- Methyl Binding Domain Proteins MBDs
- Methylation and Learning and Memory Disorders
- Histone methylation
- Histone 3 Lysine 4 Trimethylation and Memory Formation
- Histone 3 Lysine 9 Dimethylation and Memory Formation
- Histone Methylation and Other Epigenetic Modifications
- Histone Acetylation
- Histone acetyltransferases HATs
- Chromatin Remodeling
- Acetylation as an epigenetic marker
- Acetlylation as a mechanism for learning and memory
- ERKMAPK Cascade
- Long Term Potentiation
- HDACs Role in CREB CBP Dependent Transcriptional Activation
- HDAC2
- HDAC3
- HDACs Role in CNS disorders
- References
DNA Methylation
DNA methylation involves the addition of a methyl group to a 5' cytosine residue. This usually occurs at cytosines that form part of a cytosine-guanine dinucleotide (CpG sites). Methylation can lead to activation or repression of gene transcription and is mediated through the activity of DNA methyltransferases (DNMTs). DNMT3A and DNMT3B regulate de novo methylation of CpG sites, while DNMT1 maintains established methylation patterns. S-adenosyl methionine acts as the methyl donor.
The current hypothesis for how DNA methylation contributes to the storage of memories is that dynamic DNA methylation changes occur temporally to activate transcription of genes that encode for proteins whose role is to stabilize memory.
DNMTs and Memory
Miller and Sweatt demonstrated that rats trained in a contextual fear-conditioning paradigm had elevated levels of mRNA for DNMT3a and DNMT3b in the hippocampus. Fear conditioning is an associative memory task where a context, like a room, is paired with an aversive stimulus, like a foot shock; animals who have learned the association show higher levels of freezing behavior when exposed to the context even in the absence of the aversive stimulation. However, when rats were treated with the DNMT inhibitors zebularine or 5-aza-2′-deoxycytidine immediately after fear-conditioning, they demonstrated reduced learning (freezing behavior). When treated rats were re-trained 24 hours later, they performed as well as non-treated rats. Furthermore, it was shown that when these DNMT inhibitors were given 6 hours after training, and the rats were tested 24 hours later, the rats displayed normal fear memory, indicating that DNMTs are involved specifically in memory consolidation. These findings reveal the importance of dynamic changes in methylation status in memory formation.
Feng et al. created double conditional knock out (DKO) mice for the genes DNMT3a and DNMT1. These mice were shown to have significantly weakened long-term potentiation (LTP) and much more easily stimulated long-term depression (LTD) in the hippocampus. When tested in the Morris water navigation task, which is used to study hippocampus-dependent spatial memory, the DNMT3a/DNMT1 DKO mice took longer to find the platform than control mice. Single knock-out mice (SKO) for either DNMT3a or DNMT1 performed normally. DKO mice were also unable to consolidate memory after fear-conditioning. Since SKO mice did not exhibit the same learning and memory defects as the DKO mice, it was concluded that DNMT3a and DNMT1 play redundant roles in regulating learning and memory.
When DNMTs are inhibited in the prefrontal cortex, recall of existing memories is impaired, but not the formation of new ones. This indicates that DNA methylation may be circuit-specific when it comes to regulating the formation and maintenance of memories.
DNA Methylation Targets
The memory suppressor gene, protein phosphatase 1 (PP1), was shown to have increased CpG island methylation after contextual-fear conditioning. This corresponded to decreased levels of PP1 mRNA in the hippocampus of the trained rats. When DNMTs were inhibited, increased methylation at the PP1 gene was no longer observed. These data suggest that during memory consolidation in associative learning tasks, CpG methylation is used to inhibit the expression of PP1, a gene that negatively inhibits memory formation.
Demethylation and Memory
While DNA methylation is necessary to inhibit genes involved in memory suppression, DNA demethylation is important in activating genes whose expression is positively correlated with memory formation. Sweatt and Miller also showed that the gene reelin, which is involved in long term potentiation induction, had a reduced methylation profile and increased reelin mRNA in fear-conditioned versus control rats. Brain-derived neurotrophic factor (BDNF), another important gene in neural plasticity, has also been shown to have reduced methylation and increased transcription in animals that have undergone learning. While these studies have been linked to the hippocampus, recent evidence has also shown increased demethylation of reelin and BDNF in the medial prefrontal cortex (mPFC), an area involved in cognition and emotion.
The mechanism behind this experience-dependent demethylation response is not fully understood, although some evidence shows that DNMTs may be involved in this as well. It has also been suggested that members of the DNA damage repair GADD45 family may contribute to this demethylation process.
Methyl-Binding Domain Proteins (MBDs)
Mice that have genetic disruptions for CpG binding protein 2 (MeCP2) have been shown to have significant problems in hippocampus-dependent memory and have impaired hippocampal LTP.
Methylation and Learning and Memory Disorders
Changes in expression of genes associated with post-traumatic stress disorder (PTSD), which is characterized by an impaired extinction of traumatic memory, may be mediated by DNA methylation. In schizophrenics, it has been shown that reelin is down-regulated through increased DNA methylation at promoter regions in GABAergic interneurons. DNMT1 has also been shown to be upregulated in these cells.
Histone methylation
Methylation of histones may either increase or decrease gene transcription depending on which histone is modified, the amino acid that is modified, and the number of methyl groups added. In the case of lysine methylation, three types of modifications exist: monomethylated, dimethylated, or trimethylated lysines. The di- or trimethylation of histone H3 at lysine 9 (H3K9) has been associated with transcriptionally silent regions, while the di- or trimethylation of histone H3 at lysine 4 (H3K4) is associated with transcriptionally active genes.
Histone 3 Lysine 4 Trimethylation and Memory Formation
The hippocampus is an important brain region in memory formation. H3K4 trimethylation is associated with active transcription. In contextual fear conditioning experiments in rats, it was found that levels of H3K4 trimethylation increases in the hippocampus after fear conditioning. In these experiments by Gupta et al., a connection was made between changes in histone methylation and active gene expression during the consolidation of associative memories. In this same study, it was also found that these histone methylations were reversible, as the levels of trimethylation of H3K4 returned to basal levels after a period of 24 hours. This indicated that active demethylation was occurring following memory consolidation. To further explore the role of methyltransferases in long-term memory formation, this study applied the same fear conditioning tests on rats deficient in Mll, a H3K4-specific methyltransferase. The rats with a heterozygous mutant Mll+/- gene showed a significant reduction in their ability to form long-term memories compared to normal rats with an intact Mll gene. Therefore, H3K4 methyltransferases, such as Mll, must have an essential role in long-term memory formation in the hippocampus.
The change in methylation state of histones at the location of specific gene promoters, as opposed to just genome-wide, is also involved in memory formation. Zif268 and BDNF genes are critical for memory consolidation. H3K4 trimethylation increases around both of the Zif268 and BDNF promoters following contextual fear conditioning, when these genes are transcriptionally active. This demonstrates that at the time of memory consolidation, the transcription of memory formation genes such as Zif268 and bdnf is regulated by histone methylation.
Histone 3 Lysine 9 Dimethylation and Memory Formation
Histone H3 lysine 9 dimethylation is associated with transcriptional silencing. The G9a/G9a-like protein (GLP) complex is a methyltransferase specific for producing this modification. One study examined the role of G9a/GLP-mediated transcriptional silencing in the hippocampus and entorhinal cortex (EC) during memory consolidation. It was found that the inhibition of G9a/GLP in the EC, but not in the hippocampus, results in the enhancement of long-term memory formation. In addition, G9a/GLP inhibition in the entorhinal cortex altered histone H3 lysine 9 dimethylation in the Cornu Ammonis area 1 of the hippocampus, suggesting the importance of this complex in mediating connectivity between these two brain regions. Therefore, the G9a/GLP complex plays an important role in histone methylation and long term memory formation in the hippocampus and the EC.
Histone Methylation and Other Epigenetic Modifications
Histone methylation marks are also correlated with other epigenetic modifications, such as histone deacetylation and DNA methylation, in the context of learning and memory. Reduced histone deacetylation is correlated with an increase in H3K9 dimethylation, a modification associated with transcriptional silencing. Therefore, histone deacetylase inhibitors may be applied to increase histone acetylation and suppress H3K9 dimethylation, thereby increasing gene transcription. In the case of DNA methylation, it was found that increases in H3K4 trimethylation correlate with altered DNA methylation of CpG sites at the promoter of Zif268, a gene involved in memory formation, after fear conditioning. Gupta et al. showed that DNA methylation at the Zif268 promoter increased after fear conditioning, correlating with an increase in Zif268 gene expression. This finding was surprising, since it was previously thought that DNA methylation resulted in transcriptional silencing.
Histone Acetylation
Acetylation involves the replacement of a hydrogen with an acetyl group. In a biological context, acetylation is most often associated with the modification of proteins, specifically histones. The acetylation reaction is most often catalyzed by enzymes that contain histone acetyltransferase (HAT) activity.
Histone acetyltransferases (HATs)
HATs are enzymes responsible for the acetylation of amino acids. HATs acetylate by converting the lysine side group of amino acids with the addition of an acetyl group from an acetyl CoA molecule, creating acetyl lysine. HAT enzymes are most often associated with histone proteins and work to regulate the interaction between histones and the DNA that is wrapped around them. HATs are not only restricted to the acetylation of histone but can also acetylate many other proteins implicated in the manipulation of gene expression like that of transcription factors and receptor proteins.
Chromatin Remodeling
Acetylation is one of the main mechanisms implicated in the process of chromatin remodeling. Chromatin remodeling affects the regulation of gene expression by altering the relationship between nucleosomes and DNA. Acetylation of histones removes positive charge, which reduces the level of interaction between the formerly positively charged histone and the negatively charged phosphate groups of the DNA wrapped around the nucleosome complex. This alteration in charges causes a relaxation of DNA from the nucleosome, this relaxed section is seen to have higher levels of gene expression than non acetylated regions.
Acetylation as an epigenetic marker
Patterns of histone acetylation have been useful as a source of epigenetic information due to their ability to reflect changes in transcription rates and the maintenance of gene expression patterns. This acetylation code can then be read and provide generous information for the study of inheritance patterns of epigenetic changes like that of learning, memory and disease states.
Acetlylation as a mechanism for learning and memory
The role of epigenetic mechanisms and chromatin remodeling has been implicated in both synaptic plasticity and neuronal gene expression. Studies with histone deactylase complex inhibitors like SAHA, toluene, garcinol, trichostatin A and sodium butyrate have shown that acetylation is important for the synaptic plasticity of the brain; by inhibiting deactylase complexes total acetylation rates in the brain increased leading to increased rates of transcription and enhanced memory consolidation. By using various learning assays like the Morris water maze test and fear conditioning assays in conjunction with acetlyation influencing drugs it was shown that acetylation patterns in the hippocampus are integral to memory association and learning behavior. Studies with various HDAC inhibitors and neural development have shown increased learning and memory, as a result of an increased acetylation state. Conversely studies conducted with HAT inhibitors yielded impairment of memory consolidation and an overall decrease in learning.
ERK/MAPK Cascade
Studies have shown that the ERK/MAPK cascade is important for the regulation of lysine acetylation in the insular cortex of the brain (A part of the brain implicated in the formation of taste memories). The activation of the ERK/MAPK cascade was seen in mice after the introduction of a new taste, the cascade was shown to be necessary for the memory of the taste to be formed. The proposed mechanism for how this cascade works is that MAPK regulates histone acetylation and subsequent chromatin remodeling by means of downstream effectors, such as the CREB binding protein (which has HAT activity). By observing the rates of acetylation in the insular cortex researchers were able to determine which patterns of acetylation were due to deacetylase or acetylase activity and which were a result of lysine acetyltransferase activity.
Long Term Potentiation
Long term potentiation (LTP) is the enhancement of signal strength between neurons. LTP is the basis of synaptic plasticity and plays a pivotal role in memory formation. LTP is dependent on the activity of NMDA receptors in the brain and it has been shown that NMDA activity influences acetylation. When NMDA receptors are activated they cause an influx of calcium into the cell which in turn activates various signal pathways that ultimately activate the ERK pathway which then modulates transcription factors like CREB. CREB then recruits a HAT to help create and stabilize the long term formation of memory, often through the self-perpetuation of acetylated histones. Studies done on Acetylation of histone H3 in the CA1 region of the hippocampus show that the activation of NMDA receptors increased the acetylation of H3 and conversely inhibition of the ERK pathway in the CA1 region resulted in a decrease in acetylation of H3. In summary:
HDACs' Role in CREB: CBP-Dependent Transcriptional Activation
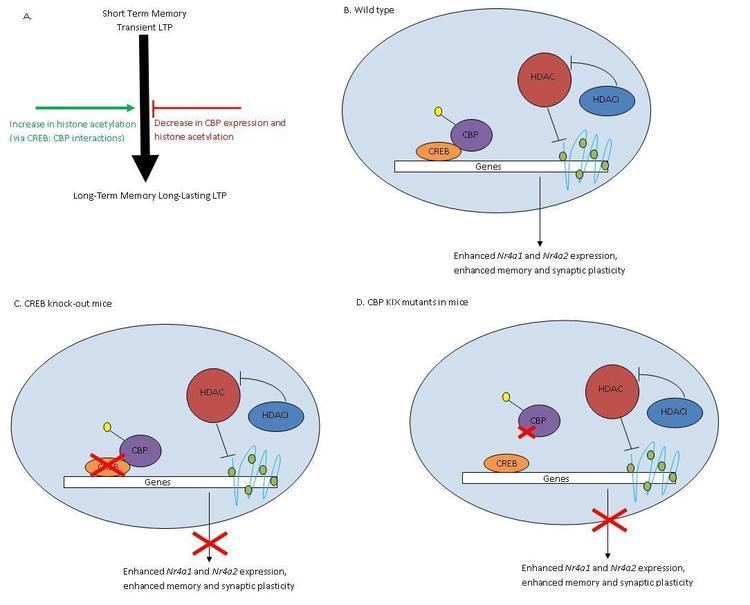
Histone deacetylases (HDAC) remove acetyl groups (-COCH3) from histones altering chromatin structures and decreasing accessibility of transcriptional factors to DNA, thereby reducing transcription of genes. HDACs have shown to play a role in learning and memory through their regulation in the CREB-CBP pathway.
Studies conclude that HDAC inhibitors such as trichostatin A (TSA) increase histone acetylation and improve synaptic plasticity and long-term memory (Fig 1A). CREB, a cAMP response element-binding protein and transcriptional activator, binds CBP forming the CREB: CBP complex. This complex activates genes involved in synaptic formation and long-term memory.(Fig 1B) TSA treatments in the hippocampal CA1 region of mice increased acetylation levels and enhanced long-term potentiation (LTP), a mechanism involved in learning and memory (Fig 1B). However, TSA treatments in CBP mutants lacking KIX domains did not effect LTP in mice (Fig 1D). The KIX domain allows for interaction between CREB and CBP, so knocking out this region disrupts formation of the CREB: CBP complex. Knock outs of CREB produced similar results to those of mutant CBP mice (Fig 1C). Therefore, HDAC inhibition and CREB: CBP association are both necessary for memory development. TSA treatments showed increased expression levels of Nr4a1 and Nra2 genes while other CREB regulated genes were unaffected. HDAC inhibitors improve memory through activation of specific genes regulated by CREB: CBP complex.
HDAC2
The role of individual HDACs in learning and memory is not well understood, but HDAC2 has been shown to negatively regulate memory formation and synaptic plasticity.
Overexpression (OE) of HDAC1 and HDAC2 in mice resulted in decreased levels of acetylated lysines. After exposing these mice to context and tone-dependent fear conditioning experiments, HDAC1 OE mice did not change, but HDAC2 OE mice showed a decrease in freezing behavior, suggesting impairment in memory formation. On the other hand, mice with HDAC2 knockouts (KO) illustrated increased freezing levels compared to wild-type (WT) mice while HDAC1 displayed similar freezing behaviors to WTs. In summary, Guan et al. have shown that:
HDAC3
HDAC3 is also a negative regulator of long term potentiation formation. McQuown et al. have shown that:
HDACs’ Role in CNS disorders
Research has shown that HDACs and HATs play a crucial role in central nervous system (CNS) disorders such as Rett syndrome. Rubinstein-Tabyi syndrome causes mental retardation through possible mutations in CREB-binding protein and p300. However, enhancing expression of CREB-dependent genes or inhibition of HDAC activity partially restore LTP loss and ameliorate late LTP deficits. HDAC inhibitor like TSA may provide a possible therapy for Rubinstein-Tabyi syndrome. Other memory-deficit disorders which may involve HDAC inhibitors as potential therapy are: