
Specialty Medical genetics ICD-9-CM 335.0-335.1 | ICD-10 G12.0-G12.1 DiseasesDB 14093 32911 | |
 | ||
OMIM 253300 253550 253400 271150 | ||
Spinal muscular atrophy (SMA), also called autosomal recessive proximal spinal muscular atrophy in order to distinguish it from other conditions with similar name, is a rare neuromuscular disorder characterised by loss of motor neurons and progressive muscle wasting, often leading to early death.
Contents
- Classification
- Signs and symptoms
- Causes
- Diagnosis
- Preimplantation testing
- Prenatal testing
- Carrier testing
- Routine screening
- Treatment
- Management
- Orthopaedics
- Mobility support
- Respiratory care
- Nutrition
- Cardiology
- Mental health
- Prognosis
- Research directions
- SMN1 gene replacement
- SMN2 alternative splicing modulation
- SMN2 gene activation
- SMN stabilisation
- Neuroprotection
- Muscle restoration
- Stem cells
- Registries
- In popular culture
- References
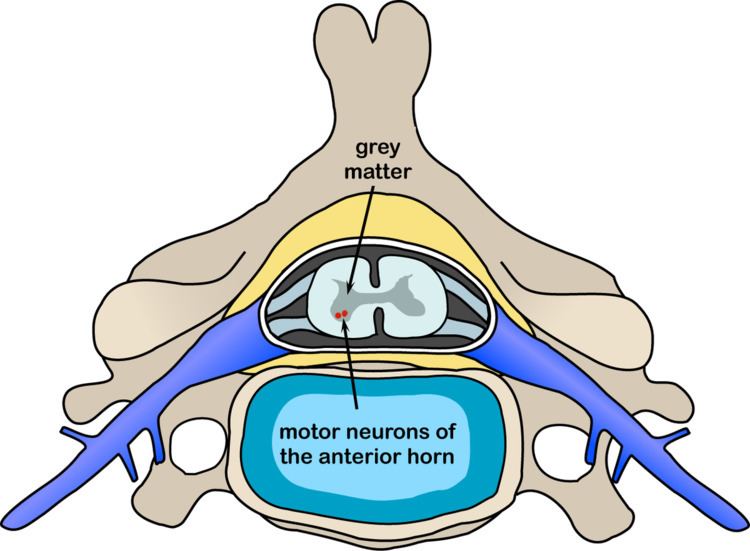
The disorder is caused by a genetic defect in the SMN1 gene, which encodes SMN, a protein widely expressed in all eukaryotic cells and necessary for survival of motor neurons. Lower levels of the protein results in loss of function of neuronal cells in the anterior horn of the spinal cord and subsequent system-wide muscle wasting (atrophy).
Spinal muscular atrophy manifests in various degrees of severity, which all have in common progressive muscle wasting and mobility impairment. Proximal muscles and lung muscles are affected first. Other body systems may be affected as well, particularly in early-onset forms of the disorder. SMA is the most common genetic cause of infant death.
Spinal muscular atrophy is an inherited disorder and is passed on in an autosomal recessive manner. In December 2016, nusinersen became the first approved drug to treat SMA while several other compounds remain in clinical trials.
Classification
SMA manifests over a wide range of severity, affecting infants through adults. The disease spectrum is variously divided into 3–5 types, in accordance either with the age of onset of symptoms or with the highest attained milestone of motor development.
The most commonly used classification is as follows:
The most severe form of SMA type I is sometimes termed SMA type 0 (or, severe infantile SMA) and is diagnosed in babies that are born so weak that they can survive only a few weeks even with intensive respiratory support. SMA type 0 should not be confused with SMARD1 which may have very similar symptoms and course but has a different genetic cause than SMA.
Motor development in people with SMA is usually assessed using validated functional scales – CHOP INTEND (The Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders) in SMA1; and either the Motor Function Measure scale or one of a few variants of Hammersmith Functional Motor Scale in SMA types 2 and 3.
The eponymous label Werdnig–Hoffmann disease (sometimes misspelled with a single n) refers to the earliest clinical descriptions of childhood SMA by Johann Hoffmann and Guido Werdnig. The eponymous term Kugelberg–Welander disease is after Erik Klas Hendrik Kugelberg (1913-1983) and Lisa Welander (1909-2001), who distinguished SMA from muscular dystrophy. Rarely used Dubowitz disease (not to be confused with Dubowitz syndrome) is named after Victor Dubowitz, an English neurologist who authored several studies on the intermediate SMA phenotype.
Signs and symptoms
The symptoms vary greatly depending on the SMA type involved, the stage of the disease, and individual factors; they commonly include:
Causes
Spinal muscular atrophy is linked to a genetic mutation in the SMN1 gene.
Human chromosome 5 contains two nearly identical genes at location 5q13: a telomeric copy SMN1 and a centromeric copy SMN2. In healthy individuals, the SMN1 gene codes the survival of motor neuron protein (SMN) which, as its name says, plays a crucial role in survival of motor neurons. The SMN2 gene, on the other hand - due to a variation in a single nucleotide (840.C→T) - undergoes alternative splicing at the junction of intron 6 to exon 8, with only 10-20% of SMN2 transcripts coding a fully functional survival of motor neuron protein (SMN-fl) and 80-90% of transcripts resulting in a truncated protein compound (SMNΔ7) which is rapidly degraded in the cell.
In individuals affected by SMA, the SMN1 gene is mutated in such a way that it is unable to correctly code the SMN protein - due to either a deletion occurring at exon 7 or to other point mutations (frequently resulting in the functional conversion of the SMN1 sequence into SMN2). Almost all people, however, have at least one functional copy of the SMN2 gene (with most having 2-4 of them) which still codes small amounts of SMN protein - around 10-20% of the normal level - allowing some neurons to survive. In the long run, however, reduced availability of the SMN protein results in gradual death of motor neuron cells in the anterior horn of spinal cord and the brain. Muscles that depend on these motor neurons for neural input now have decreased innervation (also called denervation), and therefore have decreased input from the central nervous system (CNS). Decreased impulse transmission through the motor neurons leads to decreased contractile activity of the denervated muscle. Consequently, denervated muscles undergo progressive atrophy.
Muscles of lower extremities are usually affected first, followed by muscles of upper extremities, spine and neck and, in more severe cases, pulmonary and mastication muscles. Proximal muscles are always affected earlier and to a greater degree than distal.
The severity of SMA symptoms is broadly related to how well the remaining SMN2 genes can make up for the loss of function of SMN1. This is partly related to the number of SMN2 gene copies present on the chromosome. Whilst healthy individuals carry two SMN2 gene copies, people with SMA can have anything between 1 and 4 (or more) of them, with the greater the number of SMN2 copies, the milder the disease severity. Thus, most SMA type I babies have one or two SMN2 copies; people with SMA II and III usually have at least three SMN2 copies; and people with SMA IV normally have at least four of them. However, the correlation between symptom severity and SMN2 copy number is not absolute, and there seem to exist other factors affecting the disease phenotype.
Spinal muscular atrophy is inherited in an autosomal recessive pattern, which means that the defective gene is located on an autosome. Two copies of the defective gene - one from each parent - are required to inherit the disorder: the parents may be carriers and not personally affected. SMA seems to appear de novo (i.e., without any hereditary causes) in around 2-4% of cases.
Spinal muscular atrophy affects individuals of all ethnic groups, unlike other well known autosomal recessive disorders, such as sickle cell disease and cystic fibrosis, which have significant differences in occurrence rate among ethnic groups. The overall prevalence of SMA, of all types and across all ethnic groups, is in the range of 1 per 10,000 individuals; the gene frequency is around 1:100, therefore, approximately one in 50 persons are carriers. There are no known health consequences of being a carrier. A person may learn carrier status only if one's child is affected by SMA or by having the SMN1 gene sequenced.
Affected siblings usually have a very similar form of SMA. However, occurrences of different SMA types among siblings do exist – while rare, these cases might be due to additional de novo deletions of the SMN gene, not involving the NAIP gene, or the differences in SMN2 copy numbers.
Diagnosis
Very severe SMA (type 0/1) can be sometimes evident before birth - reduction in fetal movement in the final months of pregnancy. Otherwise SMA1 manifests within the first few weeks or months of life when abnormally low muscle tone is observed in the infant (the "floppy baby syndrome").
For all SMA types,
While the above symptoms point towards SMA, the diagnosis can only be confirmed with absolute certainty through genetic testing for bi-allelic deletion of exon 7 of the SMN1 gene. Genetic testing is usually carried out using a blood sample, and MLPA is one of more frequently used gene sequencing techniques, as it also allows establishing the number of SMN2 gene copies.
Preimplantation testing
Preimplantation genetic diagnosis can be used to screen for SMA-affected embryos during in-vitro fertilisation.
Prenatal testing
Prenatal testing for SMA is possible through chorionic villus sampling, cell-free fetal DNA analysis and other methods.
Carrier testing
Those at risk of being carriers of SMN1 deletion, and thus at risk of having offspring affected by SMA, can undergo carrier analysis using a blood or saliva sample. The American College of Obstetricians and Gynecologists recommends all people thinking of becoming pregnant be tested to see if they are a carrier.
Routine screening
Routine prenatal or neonatal screening for SMA is controversial, because of the cost, and because of the severity of the disease. Some researchers have concluded that population screening for SMA is not cost-effective, at a cost of $5 million per case averted in the United States as of 2009. Others conclude that SMA meets the criteria for screening programs and relevant testing should be offered to all couples.
Treatment
Nusinersen (trade name Spinraza) is the only approved drug to treat spinal muscular atrophy. It is a proprietary antisense oligonucleotide developed by Ionis Pharmaceuticals and Biogen which is administered directly to the central nervous system using an intrathecal injection. The drug was approved by FDA in December 2016 and as of 23 December 2016 is awaiting regulatory approval in other jurisdictions.
Management
Main areas of concern are as follows:
Orthopaedics
Weak spine muscles may lead to development of kyphosis, scoliosis and other orthopaedic problems. Spine fusion is sometimes performed in people with SMA I/II once they reach the age of 8-10 to relieve the pressure of a deformed spine on the lungs. People with SMA might also benefit greatly from various forms of physiotherapy and occupational therapy.
Mobility support
Orthotic devices can be used to support the body and to aid walking. For example, orthotics such as AFO's (ankle foot orthosis) are used to stabilise the foot and to aid gait, TLSO's (thoracic lumbar sacral orthosis) are used to stabilise the torso. Assistive technologies may help in managing movement and daily activity and greatly increase the quality of life.
Respiratory care
Respiratory system requires utmost attention in SMA as once weakened it never fully recovers. Weakened pulmonary muscles in people with SMA type I/II can make breathing more difficult and pose a risk of hypoxiation, especially in sleep when muscles are more relaxed. Impaired cough reflex poses a constant risk of respiratory infection and pneumonia. Non-invasive ventilation (BiPAP) is frequently used and tracheostomy may be sometimes performed in more severe cases; both methods of ventilation prolong survival in a comparable degree, although tracheostomy prevents speech development.
Nutrition
Difficulties in jaw opening, chewing and swallowing food might put people with SMA at risk of malnutrition. A feeding tube or gastrostomy can be necessary in SMA type I and people with more severe type II. Additionally, metabolic abnormalities resulting from SMA impair β-oxidation of fatty acids in muscles and can lead to organic acidemia and consequent muscle damage, especially when fasting. It is suggested that people with SMA, especially those with more severe forms of the disease, reduce intake of fat and avoid prolonged fasting (i.e., eat more frequently than healthy people).
Cardiology
Although the heart is not a matter of routine concern, a link between SMA and certain heart conditions has been suggested.
Mental health
SMA children do not differ from the general population in their behaviour; their cognitive development can be slightly faster, and certain aspects of their intelligence are above the average. Despite their disability, SMA-affected people report high degree of satisfaction from life.
Palliative care in SMA has been standardised in the Consensus Statement for Standard of Care in Spinal Muscular Atrophy which has been recommended for standard adoption worldwide.
Prognosis
In lack of pharmacological treatment, people with SMA tend to deteriorate over time, but prognosis varies with the SMA type and disease progress which shows a great degree of individual variability.
The majority of children diagnosed with SMA type 0 and 1 do not reach the age of 4, recurrent respiratory problems being the primary cause of death. With proper care, milder SMA type 1 cases (which account for approx. 10% of all SMA1 cases) live into adulthood. Long-term survival in SMA1 is not sufficiently evidenced; however, recent advances in respiratory support seem to have brought down mortality.
In SMA type 2, the course of the disease is stable or slowly progressing and life expectancy is reduced compared to the healthy population. Death before the age of 20 is frequent, although many people with SMA live to become parents and grandparents. SMA type 3 has normal or near-normal life expectancy if standards of care are followed. Adult-onset SMA usually means only mobility impairment and does not affect life expectancy.
In all SMA types, physiotherapy has been shown to delay the progress of disease.
Research directions
Since the underlying genetic cause of SMA was identified in 1995, several therapeutic approaches have been proposed and investigated that primarily focus on increasing the availability of SMN protein in motor neurons. The main research directions are as follows:
SMN1 gene replacement
Gene therapy in SMA aims at restoring the SMN1 gene function through inserting specially crafted nucleotide sequence (a SMN1 transgene) into the cell nucleus using a viral vector; scAAV-9 and scAAV-10 are the primary viral vectors under investigation.
Only one programme has reached the clinical stage:
Work on developing gene therapy for SMA is also conducted at the Institut de Myologie in Paris and at the University of Oxford.
SMN2 alternative splicing modulation
This approach aims at modifying the alternative splicing of the SMN2 gene so that to force it to code for higher percentage of full-length SMN protein. Sometimes it is also called gene conversion, because it attempts to convert the SMN2 gene functionally into SMN1 gene.
The following splicing modulators have reached clinical stage development:
Of discontinued clinical-stage molecules, RG3039, also known as Quinazoline495, was a proprietary quinazoline derivative developed by Repligen and licensed to Pfizer in March 2014 which was discontinued shortly after, having only completed phase I trials. PTK-SMA1 was a proprietary small-molecule splicing modulator of the tetracyclines group developed by Paratek Pharmaceutical and about to enter clinical development in 2010 which however never happened. RG7800 was a molecule akin to RG7916, developed by Hoffmann-La Roche and trialled on SMA patients in 2015, whose development was put on hold indefinitely due to long-term animal toxicity.
Basic research has also identified other compounds which modified SMN2 splicing in vitro, like sodium orthovanadate and aclarubicin. Morpholino-type antisense oligonucleotides, with the same cellular target as nusinersen, remain a subject of intense research, including at the University College London and at the University of Oxford.
SMN2 gene activation
This approach aims at increasing expression (activity) of the SMN2 gene, thus increasing the amount of full-length SMN protein available.
A few compounds initially showed promise but failed to demonstrate efficacy in clinical trials:
Compounds which increased SMN2 activity in vitro but did not make it to the clinical stage include growth hormone, various histone deacetylase inhibitors, benzamide M344, hydroxamic acids (CBHA, SBHA, entinostat, panobinostat, trichostatin A, vorinostat), prolactin as well as natural polyphenol compounds like resveratrol and curcumin. Celecoxib, a p38 pathway activator, is sometimes used off-label by people with SMA based on a single animal study but such use is not backed by clinical-stage research.
SMN stabilisation
SMN stabilisation aims at stabilising the SMNΔ7 protein, the short-lived defective protein coded by the SMN2 gene, so that it is able to sustain neuronal cells.
No compounds have been taken forward to the clinical stage. Aminoglycosides showed capability to increase SMN protein availability in two studies. Indoprofen offered some promise in vitro.
Neuroprotection
Neuroprotective drugs aim at enabling the survival of motor neurons even with low levels of SMN protein.
Of clinically studied compounds which did not show efficacy, thyrotropin-releasing hormone (TRH) held some promise in an open-label uncontrolled clinical trial but did not prove effective in a subsequent double-blind placebo-controlled trial. Riluzole, a drug that has mild clinical benefit in amyotrophic lateral sclerosis, was proposed to be similarly tested in SMA, however a 2008–2010 trial in SMA types 2 and 3 was stopped early due to lack of satisfactory results.
Compounds that had some neuroprotective effect in in vitro research but never moved to in vivo studies include β-lactam antibiotics (e.g., ceftriaxone) and follistatin.
Muscle restoration
This approach aims to counter the effect of SMA by targeting the muscle tissue instead of neurons.
Stem cells
As of 2016, there has been no significant breakthrough in stem cell therapy in SMA. An experimental programme to develop a stem cell based therapeutic product for SMA was run, with financial support from the SMA community, by a US company California Stem Cell starting from 2005. It was discontinued in 2010, unable to enter the clinical stage, and the company ceased to exist shortly after.
In 2013–2014, a small number of SMA1 children in Italy received court-mandated stem cell injections following the Stamina scam, but the treatment was reported having no effect.
Whilst stem cells never form a part of any recognised therapy for SMA, a number of private companies, usually located in countries with lax regulatory oversight, take advantage of media hype and market stem cell injections as a "cure" for a vast range of disorders, including SMA. The medical consensus is that such procedures offer no clinical benefit whilst carrying significant risk, therefore people with SMA are advised against them.
Registries
People with SMA in the European Union can participate in clinical research by entering their details into registries managed by TREAT-NMD.