
 | ||
Fatty acid metabolism consists of catabolic processes that generate energy, and anabolic processes that create biologically important molecules (triglycerides, phospholipids, second messengers, local hormones and ketone bodies). Fatty acids are a family of molecules classified within the lipid macronutrient class. One role of fatty acids within animal metabolism is energy production, captured in the form of adenosine triphosphate (ATP). When compared to other macronutrient classes (carbohydrates and protein), fatty acids yield the most ATP on an energy per gram basis, when they are completely oxidized to CO2 and water by β-oxidation and the citric acid cycle. Fatty acids (mainly in the form of triglycerides) are therefore the foremost storage form of fuel in most animals, and to a lesser extent in plants. In addition, fatty acids are important components of the phospholipids that form the phospholipid bilayers out of which all the membranes of the cell are constructed (the cell wall, and the membranes that enclose all the organelles within the cells, such as the nucleus, the mitochondria, endoplasmic reticulum, and the Golgi apparatus). Fatty acids can also be cleaved, or partially cleaved, from their chemical attachments in the cell membrane to form second messengers within the cell, and local hormones in the immediate vicinity of the cell. The prostaglandins made from arachidonic acid stored in the cell membrane, are probably the most well known group of these local hormones.
Contents
- Fatty acid catabolism
- Fatty acids as an energy source
- Animals and plants synthesize carbohydrates from both glycerol and fatty acids
- Intracellular signaling
- Eicosanoid paracrine hormones
- Fatty acid synthesis
- Glycolytic end products are used in the conversion of carbohydrates into fatty acids
- Regulation of fatty acid synthesis
- Disorders
- References
Fatty acid catabolism
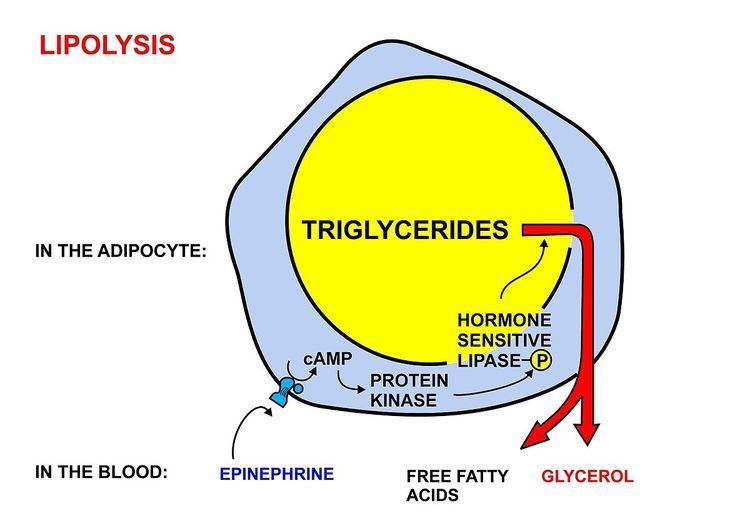
Fatty acids are released, between meals, from the fat depots in adipose tissue, where they are stored as triglycerides, as follows:
- Acyl-CoA is transferred to the hydroxyl group of carnitine by carnitine palmitoyltransferase I, located on the cytosolic faces of the outer and inner mitochondrial membranes.
- Acyl-carnitine is shuttled inside by a carnitine-acylcarnitine translocase, as a carnitine is shuttled outside.
- Acyl-carnitine is converted back to acyl-CoA by carnitine palmitoyltransferase II, located on the interior face of the inner mitochondrial membrane. The liberated carnitine is shuttled back to the cytosol, as an acyl-CoA is shuttled into the matrix.
- Dehydrogenation by acyl-CoA dehydrogenase, yielding 1 FADH2
- Hydration by enoyl-CoA hydratase
- Dehydrogenation by 3-hydroxyacyl-CoA dehydrogenase, yielding 1 NADH + H+
- Cleavage by thiolase, yielding 1 acetyl-CoA and a fatty acid that has now been shortened by 2 carbons (forming a new, shortened acyl-CoA)
Fatty acids as an energy source
Fatty acids, stored as triglycerides in an organism, are an important source of energy because they are both reduced and anhydrous. The energy yield from a gram of fatty acids is approximately 9 kcal (37 kJ), compared to 4 kcal (17 kJ) for carbohydrates. Since the hydrocarbon portion of fatty acids is hydrophobic, these molecules can be stored in a relatively anhydrous (water-free) environment. Carbohydrates, on the other hand, are more highly hydrated. For example, 1 g of glycogen can bind approximately 2 g of water, which translates to 1.33 kcal/g (4 kcal/3 g). This means that fatty acids can hold more than six times the amount of energy per unit of storage mass. Put another way, if the human body relied on carbohydrates to store energy, then a person would need to carry 31 kg (67.5 lb) of hydrated glycogen to have the energy equivalent to 4.6 kg (10 lb) of fat.
Hibernating animals provide a good example for utilizing fat reserves as fuel. For example, bears hibernate for about 7 months, and, during this entire period, the energy is derived from degradation of fat stores. Migrating birds similarly build up large fat reserves before embarking on their intercontinental journeys.
Thus the young adult human’s fat stores average between about 10–20 kg, but varies greatly depending on age, gender, and individual disposition. By contrast the human body stores only about 400 g of glycogen, of which 300 g is locked inside the skeletal muscles and is unavailable to the body as a whole. The 100 g or so of glycogen stored in the liver is depleted within one day of starvation. Thereafter the glucose that is released into the blood by the liver for general use by the body tissues, has to be synthesized from the glucogenic amino acids and a few other gluconeogenic substrates, which do not include fatty acids.
Animals and plants synthesize carbohydrates from both glycerol and fatty acids
Fatty acids are broken down to acetyl-CoA by means of beta oxidation inside the mitochondria, whereas fatty acids are synthesized from acetyl-CoA outside the mitochondria, in the cytosol. The two pathways are distinct, not only in where they occur, but also in the reactions that occur, and the substrates that are used. The two pathways are mutually inhibitory, preventing the acetyl-CoA produced by beta-oxidation from entering the synthetic pathway via the acetyl-CoA carboxylase reaction. It can also not be converted to pyruvate as the pyruvate dehydrogenase complex reaction is irreversible. Instead the acetyl-CoA produced by the beta-oxidation of fatty acids condenses with oxaloacetate, to enter the citric acid cycle. During each turn of the cycle, two carbon atoms leave the cycle as CO2 in the decarboxylation reactions catalyzed by isocitrate dehydrogenase and alpha-ketoglutarate dehydrogenase. Thus each turn of the citric acid cycle oxidizes an acetyl-CoA unit while regenerating the oxaloacetate molecule with which the acetyl-CoA had originally combined to form citric acid. The decarboxylation reactions occur before malate is formed in the cycle. Only plants possess the enzymes to convert acetyl-CoA into oxaloacetate from which malate can be formed to ultimately be converted to glucose.
However acetyl-CoA can be converted to acetoacetate, which can decarboxylate to acetone (either spontaneously, or by acetoacetate decarboxylase). It can then be further metabolized to isopropanol which is excreted in breath/urine, or by CYP2E1 into hydroxyacetone (acetol). Acetol can be converted to propylene glycol. This converts to formate and acetate (the latter converting to glucose), or pyruvate (by two alternative enzymes), or propionaldehyde, or to L-lactaldehyde then L-lactate (the common lactate isomer). Another pathway turns acetol to methylglyoxal, then to pyruvate, or to D-lactaldehyde (via S-D-lactoyl-glutathione or otherwise) then D-lactate. D-lactate metabolism (to glucose) is slow or impaired in humans, so most of the D-lactate is excreted in the urine; thus D-lactate derived from acetone can contribute significantly to the metabolic acidosis associated with ketosis or isopropanol intoxication. L-Lactate can complete the net conversion of fatty acids into glucose. The first experiment to show conversion of acetone to glucose was carried out in 1951. This, and further experiments used carbon isotopic labelling. Up to 11% of the glucose can be derived from acetone during starvation in humans.
The glycerol released into the blood during the lipolysis of triglycerides in adipose tissue can only be taken up by the liver. Here it is converted into glycerol 3-phosphate by the action of glycerol kinase which hydrolyzes one molecule of ATP per glycerol molecule which is phosphorylated. Glycerol 3-phosphate is then oxidized to dihydroxyacetone phosphate, which is, in turn, converted into glyceraldehyde 3-phosphate by the enzyme triose phosphate isomerase. From here the three carbon atoms of the original glycerol can be oxidized via glycolysis, or converted to glucose via gluconeogenesis.
Intracellular signaling
Fatty acids are an integral part of the phospholipids that make up the bulk of the plasma membranes, or cell membranes, of cells. These phospholipids can be cleaved into diacylglycerol (DAG) and inositol trisphosphate (IP3) through hydrolysis of the phospholipid, phosphatidylinositol 4,5-bisphosphate (PIP2), by the cell membrane bound enzyme phospholipase C (PLC).
An example of a diacyl-glycerol shown on the right. This DAG is 1-palmitoyl-2-oleoyl-glycerol, which contains side-chains derived from palmitic acid and oleic acid. Diacylglycerols can also have many other combinations of fatty acids attached at either the C-1 and C-2 positions or the C-1 and C-3 positions of the glycerol molecule. 1,2 disubstituted glycerols are always chiral, 1,3 disubstituted glycerols are chiral if the substituents are different from each other.
Inositol trisphosphate (IP3) functions as an intracellular second messenger, which initiates the intracellular release of calcium ions (which activates intracellular enzymes, causes the release of hormones and neurotransmitters from the cells in which they are stored, and causes smooth muscle contraction when released by IP3), and the activation of protein kinase C (PKC), which is then translocated from the cell cytoplasm to the cell membrane. Although inositol trisphosphate, (IP3), diffuses into the cytosol, diacylglycerol (DAG) remains within the plasma membrane, due to its hydrophobic properties. IP3 stimulates the release of calcium ions from the smooth endoplasmic reticulum, whereas DAG is a physiological activator of protein kinase C (PKC), promoting its translocation from the cytosol to the plasma membrane. PKC is a multifunctional protein kinase which phosphorylates serine and threonine residues in many target proteins. However PKC is only active in the presence of calcium ions, and it is DAG that increases the affinity of PKC for Ca2+ and thereby renders it active at the physiological intracellular levels of this ion.
Diacylglycerol and IP3 act transiently because both are rapidly metabolized. This is important as their message function should not linger after the message has been” received” by their target molecules. DAG can be phosphorylated to phosphatidate or it can be it can be hydrolysed to glycerol and its constituent fatty acids. IP3 is rapidly converted to into derivatives that that do not open calcium ion channels.
Eicosanoid paracrine hormones
The prostaglandins are a group of physiologically active lipid compounds having diverse hormone-like effects in animals. Prostaglandins have been found in almost every tissue in humans and other animals. They are enzymatically derived from arachidonic acid a 20-carbon polyunsaturated fatty acid. Every prostaglandin therefore contains 20 carbon atoms, including a 5-carbon ring. They are a subclass of eicosanoids and form the prostanoid class of fatty acid derivatives.
The prostaglandins are synthesized in the cell membrane by the cleavage of arachidonate from the phospholipids that make up the membrane. This is catalyzed either by phospholipase A2 acting directly on a membrane phospholipid, or by a lipase acting on DAG (diacyl-glycerol). The arachidonate is then acted upon by the cyclooxygenase component of prostaglandin synthase. This forms a cyclopentane ring in roughly the middle of the fatty acid chain. The reaction also adds 4 oxygen atoms derived from two molecules of O2. The resulting molecule is prostaglandin G2 which is converted by the hydroperoxidase component of the enzyme complex into prostaglandin H2. This highly unstable compound is rapidly transformed into other prostaglandins, prostacyclin and thromboxanes. These are then released into the interstitial fluids surrounding the cells that have manufactured the eicosanoid hormone.
If arachidonate is acted upon by a lipoxygenase instead of cyclooxygenase, Hydroxyeicosatetraenoic acids and leukotrienes are formed. They also act as local hormones.
Prostaglandins were originally believed to leave the cells via passive diffusion because of their high lipophilicity. The discovery of the prostaglandin transporter (PGT, SLCO2A1), which mediates the cellular uptake of prostaglandin, demonstrated that diffusion alone cannot explain the penetration of prostaglandin through the cellular membrane. The release of prostaglandin has now also been shown to be mediated by a specific transporter, namely the multidrug resistance protein 4 (MRP4, ABCC4), a member of the ATP-binding cassette transporter superfamily. Whether MRP4 is the only transporter releasing prostaglandins from the cells is still unclear.
The structural differences between prostaglandins account for their different biological activities. A given prostaglandin may have different and even opposite effects in different tissues. The ability of the same prostaglandin to stimulate a reaction in one tissue and inhibit the same reaction in another tissue is determined by the type of receptor to which the prostaglandin binds. They act as autocrine or paracrine factors with their target cells present in the immediate vicinity of the site of their secretion. Prostaglandins differ from endocrine hormones in that they are not produced at a specific site but in many places throughout the human body.
Prostaglandins have two derivatives: prostacyclins and thromboxanes. Prostacyclins are powerful locally acting vasodilators and inhibit the aggregation of blood platelets. Through their role in vasodilation, prostacyclins are also involved in inflammation. They are synthesized in the walls of blood vessels and serve the physiological function of preventing needless clot formation, as well as regulating the contraction of smooth muscle tissue. Conversely, thromboxanes (produced by platelet cells) are vasoconstrictors and facilitate platelet aggregation. Their name comes from their role in clot formation (thrombosis).
Fatty acid synthesis
Much like β-oxidation, straight-chain fatty acid synthesis occurs via the six recurring reactions shown below, until the 16-carbon palmitic acid is produced.
The diagrams presented show how fatty acids are synthesized in microorganisms and list the enzymes found in Escherichia coli. These reactions are performed by fatty acid synthase II (FASII), which in general contain multiple enzymes that act as one complex. FASII is present in prokaryotes, plants, fungi, and parasites, as well as in mitochondria.
In animals, as well as some fungi such as yeast, these same reactions occur on fatty acid synthase I (FASI), a large dimeric protein that has all of the enzymatic activities required to create a fatty acid. FASI is less efficient than FASII; however, it allows for the formation of more molecules, including "medium-chain" fatty acids via early chain termination.
Once a 16:0 carbon fatty acid has been formed, it can undergo a number of modifications, resulting in desaturation and/or elongation. Elongation, starting with stearate (18:0), is performed mainly in the ER by several membrane-bound enzymes. The enzymatic steps involved in the elongation process are principally the same as those carried out by FAS, but the four principal successive steps of the elongation are performed by individual proteins, which may be physically associated.
Abbreviations: ACP – Acyl carrier protein, CoA – Coenzyme A, NADP – Nicotinamide adenine dinucleotide phosphate.
Note that during fatty synthesis the reducing agent is NADPH, whereas NAD is the oxidizing agent in beta-oxidation (the breakdown of fatty acids to acetyl-CoA). This difference exemplifies a general principle that NADPH is consumed during biosynthetic reactions, whereas NADH is generated in energy-yielding reactions. (Thus NADPH is also required for the synthesis of cholesterol from acetyl-CoA; while NADH is generated during glycolysis.) The source of the NADPH is two-fold. When malate is oxidatively decarboxylated by “NADP+-linked malic enzyme" pyruvate, CO2 and NADPH are formed. NADPH is also formed by the pentose phosphate pathway which converts glucose into ribose, which can be used in synthesis of nucleotides and nucleic acids, or it can be catabolized to pyruvate.
Glycolytic end products are used in the conversion of carbohydrates into fatty acids
In humans, fatty acids are formed from carbohydrates predominantly in the liver and adipose tissue, as well as in the mammary glands during lactation. The cells of the central nervous system probably also make most of the fatty acids needed for the phospholipids of their extensive membranes from glucose, as blood-born fatty acids cannot cross the blood brain barrier to reach these cells. However, how the essential fatty acids, which mammals cannot synthesize themselves, but are nevertheless important components of cell membranes (and other functions described above) reach them is unknown.
The pyruvate produced by glycolysis is an important intermediary in the conversion of carbohydrates into fatty acids and cholesterol. This occurs via the conversion of pyruvate into acetyl-CoA in the mitochondrion. However, this acetyl CoA needs to be transported into cytosol where the synthesis of fatty acids and cholesterol occurs. This cannot occur directly. To obtain cytosolic acetyl-CoA, citrate (produced by the condensation of acetyl CoA with oxaloacetate) is removed from the citric acid cycle and carried across the inner mitochondrial membrane into the cytosol. There it is cleaved by ATP citrate lyase into acetyl-CoA and oxaloacetate. The oxaloacetate is returned to mitochondrion as malate (and then converted back into oxaloacetate to transfer more acetyl-CoA out of the mitochondrion). The cytosolic acetyl-CoA is carboxylated by acetyl CoA carboxylase into malonyl CoA, the first committed step in the synthesis of fatty acids.
Regulation of fatty acid synthesis
Acetyl-CoA is formed into malonyl-CoA by acetyl-CoA carboxylase, at which point malonyl-CoA is destined to feed into the fatty acid synthesis pathway. Acetyl-CoA carboxylase is the point of regulation in saturated straight-chain fatty acid synthesis, and is subject to both phosphorylation and allosteric regulation. Regulation by phosphorylation occurs mostly in mammals, while allosteric regulation occurs in most organisms. Allosteric control occurs as feedback inhibition by palmitoyl-CoA and activation by citrate. When there are high levels of palmitoyl-CoA, the final product of saturated fatty acid synthesis, it allosterically inactivates acetyl-CoA carboxylase to prevent a build-up of fatty acids in cells. Citrate acts to activate acetyl-CoA carboxylase under high levels, because high levels indicate that there is enough acetyl-CoA to feed into the Krebs cycle and produce energy.
High plasma levels of insulin in the blood plasma (e.g. after meals) cause the dephosphorylation of acetyl-CoA carboxylase, thus promoting the formation of malonyl-CoA from acetyl-CoA, and consequently the conversion of carbohydrates into fatty acids, while epinephrine and glucagon (released into the blood during starvation and exercise) cause the phosphorylation of this enzyme, inhibiting lipogenesis in favor of fatty acid oxidation via beta-oxidation.
Disorders
Disorders of fatty acid metabolism can be described in terms of, for example, hypertriglyceridemia (too high level of triglycerides), or other types of hyperlipidemia. These may be familial or acquired.
Familial types of disorders of fatty acid metabolism are generally classified as inborn errors of lipid metabolism. These disorders may be described as fatty oxidation disorders or as a lipid storage disorders, and are any one of several inborn errors of metabolism that result from enzyme defects affecting the ability of the body to oxidize fatty acids in order to produce energy within muscles, liver, and other cell types.