
Entrez 1374 | Ensembl ENSG00000110090 | |
 | ||
Aliases CPT1A, carnitine palmitoyltransferase 1A (liver), CPT1, CPT1-L, L-CPT1, carnitine palmitoyltransferase 1A External IDs MGI: 1098296 HomoloGene: 1413 GeneCards: CPT1A | ||
Carnitine palmitoyltransferase ii deficiency medical condition
Carnitine palmitoyltransferase I (CPT1) also known as carnitine acyltransferase I, CPTI, CAT1, CoA:carnitine acyl transferase (CCAT), or palmitoylCoA transferase I, is a mitochondrial enzyme responsible for the formation of acyl carnitines by catalyzing the transfer of the acyl group of a long-chain fatty acyl-CoA from coenzyme A to l-carnitine. The product is often Palmitoylcarnitine (thus the name), but other fatty acids may also be substrates. It is part of a family of enzymes called carnitine acyltransferases. This "preparation" allows for subsequent movement of the acyl carnitine from the cytosol into the intermembrane space of mitochondria.
Contents
- Carnitine palmitoyltransferase ii deficiency medical condition
- Streptococcus pyogenes carnitine palmitoyltransferase ii deficiency
- Structure
- Enzyme mechanism
- Biological function
- Regulation
- Clinical Significance
- Interactions
- References
Three isoforms of CPT1 are currently known: CPT1A, CPT1B, and CPT1C. CPT1 is associated with the outer mitochondrial membrane. This enzyme can be inhibited by malonyl CoA, the first committed intermediate produced during fatty acid synthesis. Its role in fatty acid metabolism makes CPT1 important in many metabolic disorders such as diabetes. Since its crystal structure is not known, its exact mechanism of action remains to be determined.
Streptococcus pyogenes carnitine palmitoyltransferase ii deficiency
Structure
CPT1 is an integral membrane protein that associates with the mitochondrial outer membrane through transmembrane regions in the peptide chain. Both the N- and C-terminal domains are exposed to the cytosolic side of the membrane.
Three isoforms of CPT1 exist in mammalian tissues. The liver isoform (CPT1A or CPTI-L) is found throughout the body on the mitochondria of all cells except for skeletal muscle cells and brown adipose cells. The muscle isoform (CPT1B or CPTI-M) is highly expressed in heart and skeletal muscle cells and brown adipose cells. A third isoform, the brain isoform (CPT1C), was isolated in 2002. It is expressed predominantly in the brain and testes. Little is known about CPT1C.
The exact structure of any of the CPT1 isoforms has not yet been determined, although a variety of in silico models for CPT1 have been created based on closely related carnitine acyltransferases, such as carnitine acetyltransferase (CRAT).
An important structural difference between CPT1 and CPT2, CRAT and carnitine octanoyltransferase (COT) is that CPT1 contains an additional domain at its N-terminal consisting of about 160 amino acids. It has been determined that this additional N-terminal domain is important for the key inhibitory molecule of CPT1, malonyl-CoA.
Two distinct binding sites have been proposed to exist in CPT1A and CPT1B. The “A site” or “CoA site” appears to bind both malonyl-CoA and palmitoyl-CoA, as well as other molecules containing coenzyme A, suggesting that the enzyme binds these molecules via interaction with the coenzyme A moiety. It has been suggested that malonyl-CoA may behave as a competitive inhibitor of CPT1A at this site. A second “O site” has been proposed to bind malonyl-CoA more tightly than the A site. Unlike the A site, the O site binds to malonyl-CoA via the dicarbonyl group of the malonate moiety of malonyl-CoA. The binding of malonyl-CoA to either the A and O sites inhibits the action of CPT1A by excluding the binding of carnitine to CPT1A. Since a crystal structure of CPT1A has yet to be isolated and imaged, its exact structure remains to be elucidated.
Enzyme mechanism
Because crystal structure data is currently unavailable, the exact mechanism of CPT1 is not currently known. A couple different possible mechanisms for CPT1 have been postulated, both of which include the histidine residue 473 as the key catalytic residue. One such mechanism based upon a carnitine acetyltransferase model is shown below in which the His 473 deprotonates carnitine while a nearby serine residue stabilizes the tetrahedral oxyanion intermediate.
A different mechanism has been proposed that suggests that a catalytic triad composed of residues Cys-305, His-473, and Asp-454 carries out the acyl-transferring step of catalysis. This catalytic mechanism involves the formation of a thioacyl-enzyme covalent intermediate with Cys-305.
Biological function
The carnitine palmitoyltransferase system is an essential step in the beta-oxidation of long chain fatty acids. This transfer system is necessary because, while fatty acids are activated (in the form of a thioester linkage to coenzyme A) on the outer mitochondrial membrane, the activated fatty acids must be oxidized within the mitochondrial matrix. Long chain fatty acids such as palmitoyl-CoA, unlike short- and medium-chain fatty acids, cannot freely diffuse through the mitochondrial inner membrane, and require a shuttle system to be transported to the mitochondrial matrix.
Carnitine palmitoyltransferase I is the first component and rate-limiting step of the carnitine palmitoyltransferase system, catalyzing the transfer of the acyl group from coenzyme A to carnitine to form palmitoylcarnitine. A translocase then shuttles the acyl carnitine across the inner mitochondrial membrane where it is converted back into palmitoyl-CoA.
By acting as an acyl group acceptor, carnitine may also play the role of regulating the intracellular CoA:acyl-CoA ratio.
Regulation
CPT1 is inhibited by malonyl-CoA, although the exact mechanism of inhibition remains unknown. The CPT1 skeletal muscle and heart isoform, CPT1B, has been shown to be 30-100-fold more sensitive to malonyl-CoA inhibition than CPT1A. This inhibition is a good target for future attempts to regulate CPT1 for the treatment of metabolic disorders.
Acetyl-CoA carboxylase (ACC), the enzyme that catalyzes the formation of malonyl-CoA from acetyl-CoA, is important in the regulation of fatty acid metabolism. Scientists have demonstrated that ACC2 knockout mice have reduced body fat and weight when compared to wild type mice. This is a result of decreased activity of ACC which causes a subsequent decrease in malonyl-CoA concentrations. These decreased malonyl-CoA levels in turn prevent inhibition of CPT1, causing an ultimate increase in fatty acid oxidation. Since heart and skeletal muscle cells have a low capacity for fatty acid synthesis, ACC may act purely as a regulatory enzyme in these cells.
Clinical Significance
The "CPT1A" form is associated with carnitine palmitoyltransferase I deficiency. This rare disorder confers risk for hepatic encephalopathy, hypoketotic hypoglycemia, seizures, and sudden unexpected death in infancy.
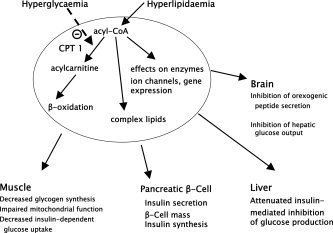
CPT1 is associated with type 2 diabetes and insulin resistance. Such diseases, along with many other health problems, cause free fatty acid (FFA) levels in humans to become elevated, fat to accumulate in skeletal muscle, and decreases the ability of muscles to oxidize fatty acids. CPT1 has been implicated in contributing to these symptoms. The increased levels of malonyl-CoA caused by hyperglycemia and hyperinsulinemia inhibit CPT1, which causes a subsequent decrease in the transport of long chain fatty acids into muscle and heart mitochondria, decreasing fatty acid oxidation in such cells. The shunting of LCFAs away from mitochondria leads to the observed increase in FFA levels and the accumulation of fat in skeletal muscle.
New research presented at the 2015 Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS) suggests that CPT1 may also play a role in the pathology of multiple sclerosis (MS). Researchers have found that mutations in the CPT1 gene appear to protect against MS in certain populations. The hypothesis is that increased lipid metabolism may help drive myelin destruction, and allowing an autoimmune attack on the nerve cells. John Dirk Nieland, PhD, and colleagues from Aalborg University in Denmark presented data showing that in animal models of MS, blocking CPT1 with a molecule called etomoxir appears to ameliorate the symptoms of MS. the researchers are now seeking support for a clinical trial in human patients.
Its importance in fatty acid metabolism makes CPT1 a potentially useful enzyme to focus on in the development of treatments of many other metabolic disorders as well.
Interactions
CPT1 is known to interact with many proteins, including ones from the NDUF family, PKC1, and ENO1.
In HIV, Vpr enhances PPARbeta/delta-induced PDK4, carnitine palmitoyltransferase I (CPT1) mRNA expression in cells. Knockdown of CPT1A by shRNA library screening inhibits HIV-1 replication in cultured Jurkat T-cells.