
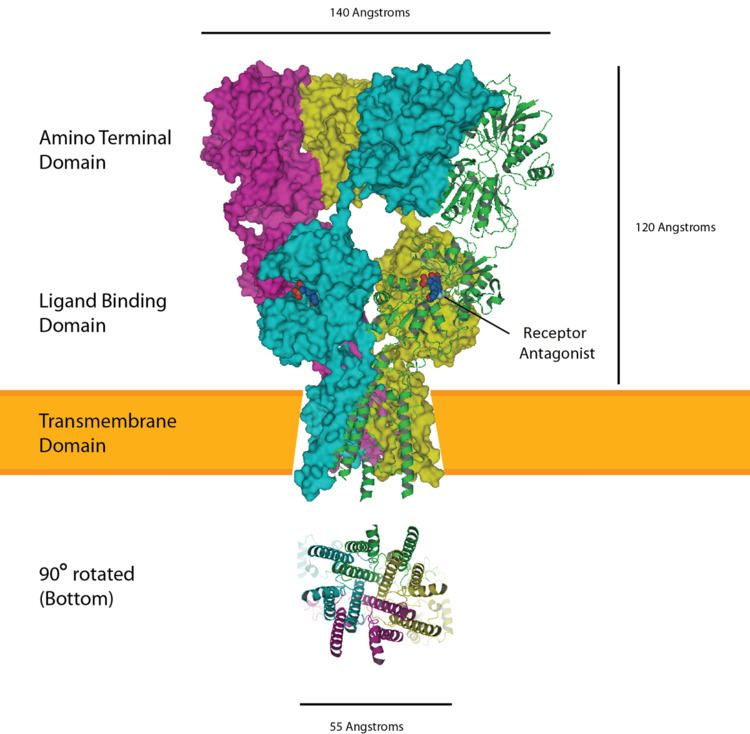
 | ||
The α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (also known as AMPA receptor, AMPAR, or quisqualate receptor) is a non-NMDA-type ionotropic transmembrane receptor for glutamate that mediates fast synaptic transmission in the central nervous system (CNS). Its name is derived from its ability to be activated by the artificial glutamate analog AMPA. The receptor was first named the "quisqualate receptor" by Watkins and colleagues after a naturally occurring agonist quisqualate and was only later given the label "AMPA receptor" after the selective agonist developed by Tage Honore and colleagues at the Royal Danish School of Pharmacy in Copenhagen. AMPARs are found in many parts of the brain and are the most commonly found receptor in the nervous system. The AMPA receptor GluA2 (GluR2) tetramer was the first glutamate receptor ion channel to be crystallized.
Contents
- Subunit composition
- Ion channel function
- Synaptic plasticity
- Molecular and signaling response to LTP inducing stimuli
- AMPA receptor trafficking to the PSD in response to LTP
- Constitutive trafficking and changes in subunit composition
- LTD induced endocytosis of AMPA receptors
- Role in Seizures
- Molecular target for epilepsy therapy
- Agonists
- Positive allosteric modulators
- Antagonists
- Negative allosteric modulators
- References
Subunit composition
AMPARs are composed of four types of subunits, designated as GluR1 (GRIA1), GluR2 (GRIA2), GluR3 (GRIA3), and GluR4, alternatively called GluRA-D2 (GRIA4), which combine to form tetramers. Most AMPARs are heterotetrameric, consisting of symmetric 'dimer of dimers' of GluR2 and either GluR1, GluR3 or GluR4. Dimerization starts in the endoplasmic reticulum with the interaction of N-terminal LIVBP domains, then "zips up" through the ligand-binding domain into the transmembrane ion pore.
The conformation of the subunit protein in the plasma membrane caused controversy for some time. While the amino acid sequence of the subunit indicated that there seemed to be four transmembrane domains (parts of the protein that pass through the plasma membrane), proteins interacting with the subunit indicated that the N-terminus seemed to be extracellular, while the C-terminus seemed to be intracellular. However, if each of the four transmembrane domains went all the way through the plasma membrane, then the two termini would have to be on the same side of the membrane. It was eventually discovered that the second "transmembrane" domain does not in fact cross the membrane at all, but kinks back on itself within the membrane and returns to the intracellular side. When the four subunits of the tetramer come together, this second membranous domain forms the ion-permeable pore of the receptor.
AMPAR subunits differ most in their C-terminal sequence, which determines their interactions with scaffolding proteins. All AMPARs contain PDZ-binding domains, but which PDZ domain they bind to differs. For example, GluR1 binds to SAP97 through SAP97's class I PDZ domain, while GluR2 binds to PICK1 and GRIP/ABP. Of note, AMPARs cannot directly bind to the common synaptic protein PSD-95 owing to incompatible PDZ domains, although they do interact with PSD-95 via stargazin (the prototypical member of the TARP family of AMPAR auxiliary subunits).
Phosphorylation of AMPARs can regulate channel localization, conductance, and open probability. GluR1 has four known phosphorylation sites at serine 818 (S818), S831, threonine 840, and S845 (other subunits have similar phosphorylation sites, but GluR1 has been the most extensively studied). S818 is phosphorylated by protein kinase C, and is necessary for long-term potentiation (LTP; for GluR1's role in LTP, see below). S831 is phosphorylated by CaMKII and PKC during LTP, which helps deliver GluR1-containing AMPAR to the synapse, and increases their single channel conductance. The T840 site was more recently discovered, and has been implicated in LTD. Finally, S845 is phosphorylated by PKA which regulates its open probability.
Ion channel function
Each AMPAR has four sites to which an agonist (such as glutamate) can bind, one for each subunit. The binding site is believed to be formed by the N-terminal tail and the extracellular loop between transmembrane domains three and four. When an agonist binds, these two loops move towards each other, opening the pore. The channel opens when two sites are occupied, and increases its current as more binding sites are occupied. Once open, the channel may undergo rapid desensitization, stopping the current. The mechanism of desensitization is believed to be due to a small change in angle of one of the parts of the binding site, closing the pore. AMPARs open and close quickly (1ms), and are thus responsible for most of the fast excitatory synaptic transmission in the central nervous system. The AMPAR's permeability to calcium and other cations, such as sodium and potassium, is governed by the GluR2 subunit. If an AMPAR lacks a GluR2 subunit, then it will be permeable to sodium, potassium, and calcium. The presence of a GluR2 subunit will almost always render the channel impermeable to calcium. This is determined by post-transcriptional modification — RNA editing — of the Q-to-R editing site of the GluR2 mRNA. Here, A→I editing alters the uncharged amino acid glutamine (Q) to the positively charged arginine (R) in the receptor's ion channel. The positively charged amino acid at the critical point makes it energetically unfavourable for calcium to enter the cell through the pore. Almost all of the GluR2 subunits in CNS are edited to the GluR2(R) form. This means that the principal ions gated by AMPARs are sodium and potassium, distinguishing AMPARs from NMDA receptors (the other main ionotropic glutamate receptors in the brain), which also permit calcium influx. Both AMPA and NMDA receptors, however, have an equilibrium potential near 0 mV. The prevention of calcium entry into the cell on activation of GluR2-containing AMPARs is proposed to guard against excitotoxicity.
The subunit composition of the AMPAR is also important for the way this receptor is modulated. If an AMPAR lacks GluR2 subunits, then it is susceptible to being blocked in a voltage-dependent manner by a class of molecules called polyamines. Thus, when the neuron is at a depolarized membrane potential, polyamines will block the AMPAR channel more strongly, preventing the flux of potassium ions through the channel pore. GluR2-lacking AMPARs are, thus, said to have an inwardly rectifying I/V curve, which means that they pass less outward current than inward current.
Alongside RNA editing, alternative splicing allows a range of functional AMPA receptor subunits beyond what is encoded in the genome. In other words, although one gene (GRIA1–GRIA4) is encoded for each subunit (GluR1–GluR4), splicing after transcription from DNA allows some exons to be translated interchangeably, leading to several functionally different subunits from each gene.
The flip/flop sequence is one such interchangeable exon. A 38-amino acid sequence found prior to (i.e., before the N-terminus of) the fourth membranous domain in all four AMPAR subunits, it determines the speed of desensitisation of the receptor and also the speed at which the receptor is resensitised and the rate of channel closing. The flip form is present in prenatal AMPA receptors and gives a sustained current in response to glutamate activation.
Synaptic plasticity
AMPA receptors (AMPAR) are both glutamate receptors and cation channels that are integral to plasticity and synaptic transmission at many postsynaptic membranes. One of the most widely and thoroughly investigated forms of plasticity in the nervous system is known as long-term potentiation, or LTP. There are two necessary components of LTP: presynaptic glutamate release and postsynaptic depolarization. Therefore, LTP can be induced experimentally in a paired electrophysiological recording when a presynaptic cell is stimulated to release glutamate on a postsynaptic cell that is depolarized. The typical LTP induction protocol involves a “tetanus” stimulation, which is a 100 Hz stimulation for 1 second. When one applies this protocol to a pair of cells, one will see a sustained increase of the amplitude of the EPSP following tetanus. This response is interesting since it is thought to be the physiological correlate for learning and memory in the cell. In fact, it was recently shown that, following a single paired-avoidance paradigm in mice, LTP could be recorded in some hippocampal synapses in vivo.
The molecular basis for LTP has been extensively studied, and AMPARs have been shown to play an integral role in the process. Both GluR1 and GluR2 play an important role in synaptic plasticity. It is now known that the underlying physiological correlate for the increase in EPSP size is a postsynaptic upregulation of AMPARs at the membrane, which is accomplished through the interactions of AMPARs with many cellular proteins.
The simplest explanation for LTP is as follows (see the long-term potentiation article for a much more detailed account). Glutamate binds to postsynaptic AMPARs and another glutamate receptor, the NMDA receptor (NMDAR). Ligand binding causes the AMPARs to open, and Na+ flows into the postsynaptic cell, resulting in a depolarization. NMDARs, on the other hand, do not open directly because their pores are occluded at resting membrane potential by Mg2+ ions. NMDARs can open only when a depolarization from the AMPAR activation leads to repulsion of the Mg2+ cation out into the extracellular space, allowing the pore to pass current. Unlike AMPARs, however, NMDARs are permeable to both Na+ and Ca2+. The Ca2+ that enters the cell triggers the upregulation of AMPARs to the membrane, which results in a long-lasting increase in EPSP size underlying LTP. The calcium entry also phosphorylates CaMKII, which phosphorylates AMPARs, increasing their single-channel conductance.
Molecular and signaling response to LTP-inducing stimuli
The mechanism for LTP has long been a topic of debate, but, recently, mechanisms have come to some consensus. AMPARs play a key role in this process, as one of the key indicators of LTP induction is the increase in the ratio of AMPAR to NMDARs following high-frequency stimulation. The idea is that AMPARs are trafficked from the dendrite into the synapse and incorporated through some series of signaling cascades.
AMPARs are initially regulated at the transcriptional level at their 5’ promoter regions. There is significant evidence pointing towards the transcriptional control of AMPA receptors in longer-term memory through cAMP response element-binding protein (CREB) and Mitogen-activated protein kinases (MAPK). Messages are translated on the rough endoplasmic reticulum (rough ER) and modified there. Subunit compositions are determined at the time of modification at the rough ER. After post-ER processing in the golgi apparatus, AMPARs are released into the perisynaptic membrane as a reserve waiting for the LTP process to be initiated.
The first key step in the process following glutamate binding to NMDARs is the influx of calcium through the NMDA receptors and the resultant activation of Ca2+/calmodulin-dependent protein kinase (CaMKII). Blocking either this influx or the activation of CaMKII prevents LTP, showing that these are necessary mechanisms for LTP. In addition, profusion of CaMKII into a synapse causes LTP, showing that it is a causal and sufficient mechanism.
CaMKII has multiple modes of activation to cause the incorporation of AMPA receptors into the perisynaptic membrane. CAMKII enzyme is eventually responsible for the development of the actin cytoskeleton of neuronal cells and, eventually, for the dendrite and axon development (synaptic plasticity). The first is direct phosphorylation of synaptic-associated protein 97(SAP97). First, SAP-97 and Myosin-VI, a motor protein, are bound as a complex to the C-terminus of AMPARs. Following phosphorylation by CaMKII, the complex moves into the perisynaptic membrane. The second mode of activation is through the MAPK pathway. CaMKII activates the Ras proteins, which go on to activate p42/44 MAPK, which drives AMPAR insertion directly into the perisynaptic membrane.
AMPA receptor trafficking to the PSD in response to LTP
Once AMPA receptors are transported to the perisynaptic region through PKA or SAP97 phosphorylation, receptors are then trafficked to the postsynaptic density (PSD). However, this process of trafficking to the PSD still remains controversial. One possibility is that, during LTP, there is lateral movement of AMPA receptors from perisynpatic sites directly to the PSD. Another possibility is that exocytosis of intracellular vesicles is responsible for AMPA trafficking to the PSD directly. Recent evidence suggests that both of these processes are happening after an LTP stimulus; however, only the lateral movement of AMPA receptors from the perisynaptic region enhances the number of AMPA receptors at the PSD. The exact mechanism responsible for lateral movement of AMPA receptors to the PSD remains to be discovered; however, research has discovered several essential proteins for AMPA receptor trafficking. For example, overexpression of SAP97 leads to increased AMPA receptor trafficking to synapses. In addition to influencing synaptic localization, SAP97 has also been found to influence AMPA receptor conductance in response to glutamate. Myosin proteins are calcium sensitive motor proteins that have also been found to be essential for AMPA receptor trafficking. Disruption of myosin Vb interaction with Rab11 and Rab11-FIP2 blocks spine growth and AMPA receptor trafficking. Therefore, it is possible that myosin may drive the lateral movement of AMPA receptors in the perisynpatic region to the PSD. Transmembrane AMPA receptor regulatory proteins (TARPs) are a family proteins that associate with AMPA receptors and control their trafficking and conductance. CACNG2 (Stargazin) is one such protein and is found to bind AMPA receptors in the perisynaptic and postsynaptic regions. The role of stargazin in trafficking between the perisynaptic and postsynaptic regions remains unclear; however, stargazin is essential for immobilizing AMPA receptors in the PSD by interacting with PSD-95. PSD-95 stabilizes AMPA receptors to the synapse and disruption of the stargazin-PSD-95 interaction suppressed synaptic transmission.
Constitutive trafficking and changes in subunit composition
AMPA receptors are continuously being trafficked (endocytosed, recycled, and reinserted) into and out of the plasma membrane. Recycling endosomes within the dendritic spine contain pools of AMPA receptors for such synaptic reinsertion. Two distinct pathways exist for the trafficking of AMPA receptors: a regulated pathway and a constitutive pathway.
In the regulated pathway, GluR1-containing AMPA receptors are trafficked to the synapse in an activity-dependent manner, stimulated by NMDA receptor activation. Under basal conditions, the regulated pathway is essentially inactive, being transiently activated only upon the induction of long-term potentiation. This pathway is responsible for synaptic strengthening and the initial formation of new memories.
In the constitutive pathway, GluR1-lacking AMPA receptors, usually GluR2-GluR3 heteromeric receptors, replace the GluR1-containing receptors in a one-for-one, activity-independent manner, preserving the total number of AMPA receptors in the synapse. This pathway is responsible for the maintenance of new memories, sustaining the transient changes resulting from the regulated pathway. Under basal conditions, this pathway is routinely active, as it is necessary also for the replacement of damaged receptors.
The GluR1 and GluR4 subunits consist of a long carboxy (C)-tail, whereas the GluR2 and GluR3 subunits consist of a short carboxy-tail. The two pathways are governed by interactions between the C termini of the AMPA receptor subunits and synaptic compounds and proteins. Long C-tails prevent GluR1/4 receptors from being inserted directly into the postsynaptic density zone (PSDZ) in the absence of activity, whereas the short C-tails of GluR2/3 receptors allow them to be inserted directly into the PSDZ. The GluR2 C terminus interacts with and binds to N-ethylmaleimide sensitive fusion protein, which allows for the rapid insertion of GluR2-containing AMPA receptors at the synapse. In addition, GluR2/3 subunits are more stably tethered to the synapse than GluR1 subunits.
LTD-induced endocytosis of AMPA receptors
Long-term depression enacts mechanisms to decrease AMPA receptor density in selected dendritic spines, dependent on clathrin and calcineurin and distinct from that of constitutive AMPAR trafficking. The starting signal for AMPAR endocytosis is an NMDAR-dependent calcium influx from low-frequency stimulation, which in turn activates protein phosphatases PP1 and calcineurin. However, AMPAR endocytosis has also been activated by voltage-dependent calcium channels, agonism of AMPA receptors, and administration of insulin, suggesting general calcium influx as the cause of AMPAR endocytosis. Blockage of PP1 did not prevent AMPAR endocytosis, but antagonist application to calcineurin led to significant inhibition of this process.
Calcineurin interacts with an endocytotic complex at the postsynaptic zone, explaining its effects on LTD. The complex, consisting of a clathrin-coated pit underneath a section of AMPAR-containing plasma membrane and interacting proteins, is the direct mechanism for reduction of AMPARs, in particular GluR2/GluR3 subunit-containing receptors, in the synapse. Interactions from calcineurin activate dynamin GTPase activity, allowing the clathrin pit to excise itself from the cell membrane and become a cytoplasmic vesicle. Once the clathrin coat detaches, other proteins can interact directly with the AMPARs using PDZ carboxyl tail domains; for example, glutamate receptor-interacting protein 1 (GRIP1) has been implicated in intracellular sequestration of AMPARs. Intracellular AMPARs are subsequently sorted for degradation by lysosomes or recycling to the cell membrane. For the latter, PICK1 and PKC can displace GRIP1 to return AMPARs to the surface, reversing the effects of endocytosis and LTD when appropriate. Nevertheless, the highlighted calcium-dependent, dynamin-mediated mechanism above has been implicated as a key component of LTD and as such may have applications to further behavioral research.
Role in Seizures
AMPA receptors play a key role in the generation and spread of epileptic seizures. Kainic acid, a convulsant that is widely used in epilepsy research induces seizures, in part, via activation of AMPA receptors
Molecular target for epilepsy therapy
The noncompetitive AMPA receptor antagonists talampanel and perampanel have been demonstrated to have activity in the treatment of adults with partial seizures, indicating that AMPA receptor antagonists represent a potential target for the treatment of epilepsy. Perampanel (trade name: Fycompa) received Marketing Authorisation Approval by the European Commission for the treatment of partial epilepsy on July 27, 2012. The drug was approved in the United States by the Food and Drug Administration (FDA) on October 22, 2012. As has been the case for most recently developed AEDs including pregabalin, lacosamide and ezogabine, the FDA recommended that perampanel be classified by the Drug Enforcement Administration (DEA) as a scheduled drug. It has been designated as a Schedule 3 controlled substance.