
 | ||
Excitotoxicity is the pathological process by which nerve cells are damaged or killed by excessive stimulation by neurotransmitters such as glutamate and similar substances. This occurs when receptors for the excitatory neurotransmitter glutamate (glutamate receptors) such as the NMDA receptor and AMPA receptor are overactivated by glutamatergic storm. Excitotoxins like NMDA and kainic acid which bind to these receptors, as well as pathologically high levels of glutamate, can cause excitotoxicity by allowing high levels of calcium ions (Ca2+) to enter the cell. Ca2+ influx into cells activates a number of enzymes, including phospholipases, endonucleases, and proteases such as calpain. These enzymes go on to damage cell structures such as components of the cytoskeleton, membrane, and DNA.
Contents
Excitotoxicity may be involved in spinal cord injury, stroke, traumatic brain injury, hearing loss (through noise overexposure or ototoxicity), and in neurodegenerative diseases of the central nervous system (CNS) such as multiple sclerosis, Alzheimer's disease, amyotrophic lateral sclerosis (ALS), Parkinson's disease, alcoholism or alcohol withdrawal and especially over-rapid benzodiazepine withdrawal, and also Huntington's disease. Other common conditions that cause excessive glutamate concentrations around neurons are hypoglycemia. Blood sugars are the primary glutamate removal method from inter-synaptic spaces at the NMDA and AMPA receptor site. Persons in excitotoxic shock must never fall into hypoglycemia. Patients should be given 5% glucose (dextrose) IV drip during excitotoxic shock to avoid a dangerous build up of glutamate around NMDA and AMPA neurons. When 5% glucose (dextrose) IV drip is not available high levels of fructose are given orally. Treatment is administered during the acute stages of excitotoxic shock along with glutamate antagonists. Dehydration should be avoided as this also contributes to the concentrations of glutamate in the inter-synaptic cleft and "status epilepticus can also be triggered by a build up of glutamate around inter-synaptic neurons".
History
The harmful effects of glutamate on the central nervous system (CNS) were first observed in 1954 by T. Hayashi, a Japanese scientist who noted that direct application of glutamate to the CNS caused seizure activity, though this report went unnoticed for several years. The toxicity of glutamate was then observed by D. R. Lucas and J. P. Newhouse in 1957, when the subcutaneous injection of monosodium glutamate into newborn mice destroyed the neurons in the inner layers of the retina. Later, in 1969, John Olney discovered that the phenomenon was not restricted to the retina, but occurred throughout the brain, and coined the term excitotoxicity. He also assessed that cell death was restricted to postsynaptic neurons, that glutamate agonists were as neurotoxic as their efficiency to activate glutamate receptors, and that glutamate antagonists could stop the neurotoxicity. Subsequent research by Mark Mattson provided evidence for the involvement of excitotoxicity in Alzheimer's disease, and other age-related neurodegenerative conditions that involve oxidative stress and cellular energy deficits.
Pathophysiology
Excitotoxicity can occur from substances produced within the body (endogenous excitotoxins). Glutamate is a prime example of an excitotoxin in the brain, and it is also the major excitatory neurotransmitter in the mammalian CNS. During normal conditions, glutamate concentration can be increased up to 1mM in the synaptic cleft, which is rapidly decreased in the lapse of milliseconds. When the glutamate concentration around the synaptic cleft cannot be decreased or reaches higher levels, the neuron kills itself by a process called apoptosis.
This pathologic phenomenon can also occur after brain injury and spinal cord injury. Within minutes after spinal cord injury, damaged neural cells within the lesion site spill glutamate into the extracellular space where glutamate can stimulate presynaptic glutamate receptors to enhance the release of additional glutamate. Brain trauma or stroke can cause ischemia, in which blood flow is reduced to inadequate levels. Ischemia is followed by accumulation of glutamate and aspartate in the extracellular fluid, causing cell death, which is aggravated by lack of oxygen and glucose. The biochemical cascade resulting from ischemia and involving excitotoxicity is called the ischemic cascade. Because of the events resulting from ischemia and glutamate receptor activation, a deep chemical coma may be induced in patients with brain injury to reduce the metabolic rate of the brain (its need for oxygen and glucose) and save energy to be used to remove glutamate actively. (The main aim in induced comas is to reduce the intracranial pressure, not brain metabolism).
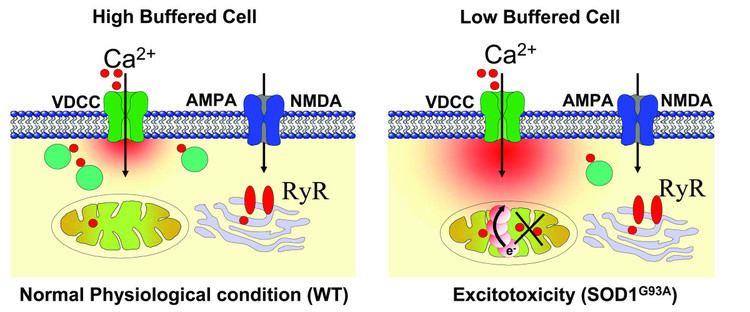
Increased extracellular glutamate levels leads to the activation of Ca2+ permeable NMDA receptors on myelin sheaths and oligodendrocytes, leaving oligodendrocytes susceptible to Ca2+ influxes and subsequent excitotoxicity. One of the damaging results of excess calcium in the cytosol is initiating apoptosis through cleaved caspase processing. Another damaging result of excess calcium in the cytosol is the opening of the mitochondrial permeability transition pore, a pore in the membranes of mitochondria that opens when the organelles absorb too much calcium. Opening of the pore may cause mitochondria to swell and release reactive oxygen species and other proteins that can lead to apoptosis. The pore can also cause mitochondria to release more calcium. In addition, production of adenosine triphosphate (ATP) may be stopped, and ATP synthase may in fact begin hydrolysing ATP instead of producing it.
Inadequate ATP production resulting from brain trauma can eliminate electrochemical gradients of certain ions. Glutamate transporters require the maintenance of these ion gradients to remove glutamate from the extracellular space. The loss of ion gradients results in not only the halting of glutamate uptake, but also the reversal of the transporters. The Na+-glutamate transporters on neurons and astrocytes can reverse their glutamate transport and start secreting glutamate at a concentration capable of inducing excitotoxicity. This results in a buildup of glutamate and further damaging activation of glutamate receptors.
On the molecular level, calcium influx is not the only factor responsible for apoptosis induced by excitoxicity. Recently, it has been noted that extrasynaptic NMDA receptor activation, triggered by both glutamate exposure or hypoxic/ischemic conditions, activate a CREB (cAMP response element binding) protein shut-off, which in turn caused loss of mitochondrial membrane potential and apoptosis. On the other hand, activation of synaptic NMDA receptors activated only the CREB pathway, which activates BDNF (brain-derived neurotrophic factor), not activating apoptosis.
Exogenous excitotoxins
Exogenous excitotoxins refer to neurotoxins that also act at postsynaptic cells but are not normally found in the body. These toxins may enter the body of an organism from the environment through wounds, food intake, aerial dispersion etc. Common excitotoxins include glutamate analogs that mimic the action of glutamate at glutamate receptors, including AMPA and NMDA receptors.
BMAA
The environmentally ubiquitous L-alanine derivative, β-methylamino-L-alanine (BMAA) has long been identified as a neurotoxin which was first associated with the amyotrophic lateral sclerosis/parkinsonism–dementia complex (ALS/PDC) in the Chamorro people of Guam. The widespread occurrence of BMAA can be attributed to cyanobacteria which produce BMAA as a result of complex reactions under nitrogen stress. Following research, excitotoxicity appears to be the likely mode of action for BMAA which acts as a glutamate agonist, activating AMPA and NMDA receptors and causing damage to cells even at relatively low concentrations of 10 μM. The subsequent uncontrolled influx of Ca2+ then leads to the pathophysiology described above. Further evidence of the role of BMAA as an excitotoxin is rooted in the ability of NMDA antagonists like MK801 to block the action of BMAA. More recently, evidence has been found that BMAA is misincorporated in place of L-serine in human proteins. It should be noted that a considerable portion of the research relating to the toxicity of BMAA has been conducted on rodents. While BMAA has been detected in brain tissue of deceased ALS/PDC patients, further insight is required to trace neurodegenerative pathology in humans to BMAA.