
Entrez 6401 | Ensembl ENSG00000007908 | |
 | ||
External IDs MGI: 98278 HomoloGene: 389 GeneCards: SELE | ||
E-selectin, also known as CD62 antigen-like family member E (CD62E), endothelial-leukocyte adhesion molecule 1 (ELAM-1), or leukocyte-endothelial cell adhesion molecule 2 (LECAM2), is a cell adhesion molecule expressed only on endothelial cells activated by cytokines. Like other selectins, it plays an important part in inflammation. In humans, E-selectin is encoded by the SELE gene.
Contents
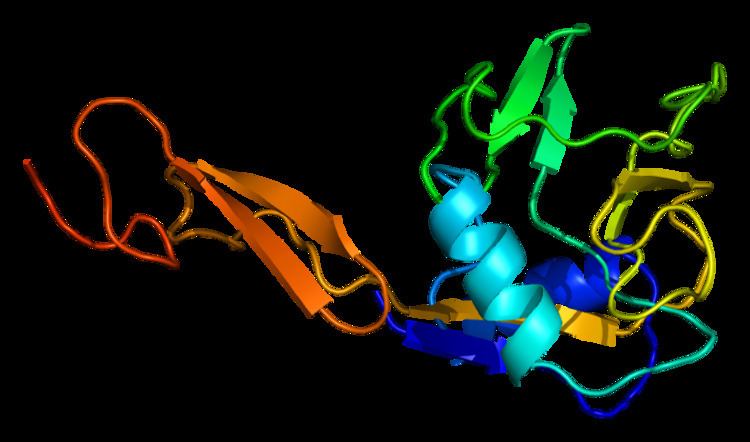
Structure
E selectin has a cassette structure: an N-terminal, C-type lectin domain, an EGF (epidermal-growth-factor)-like domain, 6 Sushi domain (SCR repeat) units, a transmembrane domain (TM) and an intracellular cytoplasmic tail (cyto). The three-dimensional structure of the ligand-binding region of human E-selectin has been determined at 2.0 Å resolution in 1994. The structure reveals limited contact between the two domains and a coordination of Ca2+ not predicted from other C-type lectins. Structure/function analysis indicates a defined region and specific amino-acid side chains that may be involved in ligand binding. The E-selectin bound to sialyl-LewisX (SLeX; NeuNAcα2,3Galβ1,4[Fucα1,3]GlcNAc) tetrasaccharide was solved in 2000.
Gene and regulation
In humans, E-selectin is encoded by the SELE gene. Its C-type lectin domain, EGF-like, SCR repeats, and transmembrane domains are each encoded by separate exons, whereas the E-selectin cytosolic domain derives from two exons. The E-selectin locus flanks the L-selectin locus on chromosome 1.
Different from P-selectin, which is stored in vesicles called Weibel-Palade bodies, E-selectin is not stored in the cell and has to be transcribed, translated, and transported to the cell surface. The production of E-selectin is stimulated by the expression of P-selectin which in turn, is stimulated by tumor necrosis factor α (TNFα), interleukin-1 (IL-1) and lipopolysaccharide (LPS). It takes about two hours, after cytokine recognition, for E-selectin to be expressed on the endothelial cell's surface. Maximal expression of E-selectin occurs around 6–12 hours after cytokine stimulation, and levels returns to baseline within 24 hours.
Shear forces are also found to affect E-selectin expression. A high laminar shear enhances acute endothelial cell response to interleukin-1β in naïve or shear-conditioned endothelial cells as may be found in the pathological setting of ischemia/reperfusion injury while conferring rapid E-selectin down regulation to protect against chronic inflammation.
Phytoestrogens, plant compounds with estrogen-like biological activity, such as genistein, formononetin, biochanin A and daidzein, as well as a mixture of these phytoestrogens were found able to reduce E-selectin as well as VCAM-1 and ICAM-1 on cell surface and in culture supernatant.
Ligands
E-selectin recognizes and binds to sialylated carbohydrates present on the surface proteins of certain leukocytes. E-selectin ligands are expressed by neutrophils, monocytes, eosinophils, memory-effector T-like lymphocytes, and natural killer cells. Each of these cell types is found in acute and chronic inflammatory sites in association with expression of E-selectin, thus implicating E-selectin in the recruitment of these cells to such inflammatory sites.
These carbohydrates include members of the Lewis X and Lewis A families found on monocytes, granulocytes, and T-lymphocytes.
The glycoprotein ESL-1, present on neutrophils and myeloid cells, was the first counter-receptor for E-selectin to be described. It is a variant of the tyrosine kinase FGF glycoreceptor, raising the possibility that its binding to E-selectin is involved in initiating signaling in the bound cells
P-selectin glycoprotein ligand-1 (PSGL-1) derived from human neutrophils is also a high-efficiency ligand for endothelium-expressed E-selectin under flow. It mediates the rolling of leukocytes on the activated endothelium surrounding an inflamed tissue.
Both ESL-1 and PSGL-1 should bear sialyl Lewis a/x in order to bind E/P-selectins.
E-selectin is found to mediate the adhesion of tumor cells to endothelial cells, by binding to E-selectin ligands on the tumor cells. E-selectin ligands also play a role in cancer metastasis. The role of these two E-selectin ligands in metastasis in vivo is poorly defined and remains to be firmly demonstrated. PSGL-1 was detected on the surfaces of bone-metastatic prostate tumor cells, suggesting that it may have a functional role in the bone tropism of prostate tumor cells.
In cancer cells, CD44, death receptor-3 (DR3), LAMP1, and LAMP2 were identified as E-selectin ligands present on colon cancer cells., and CD44v, Mac2-BP, and gangliosides were identified as E-selectin ligands present on breast cancer cells.
On human neutrophils the glycosphingolipid NeuAcα2-3Galβ1-4GlcNAcβ1-3[Galβ1-4(Fucα1-3)GlcNAcβ1-3]2[Galβ1-4GlcNAcβ1-3]2Galβ1-4GlcβCer (and closely related structures) are functional E-selectin receptors.
Role in inflammation
During inflammation, E-selectin plays an important part in recruiting leukocytes to the site of injury. The local release of cytokines IL-1 and TNF-α by Macrophages in the inflamed tissue induces the over-expression of E-selectin on endothelial cells of nearby blood vessels. Leukocytes in the blood expressing the correct ligand will bind with low affinity to E-selectin, also under the shear stress of blood flow, causing the leukocytes to "roll" along the internal surface of the blood vessel as temporary interactions are made and broken.
As the inflammatory response progresses, chemokines released by injured tissue enter the blood vessels and activate the rolling leukocytes, which are now able to tightly bind to the endothelial surface and begin making their way into the tissue.
P-selectin has a similar function, but is expressed on the endothelial cell surface within minutes as it is stored within the cell rather than produced on demand.
Role in Cancer
Cancer cells are able to infiltrate the inflammatory system by interacting with selectins. E-selectin mediates the adhesion of tumor cells to endothelial cells, by binding to E-selectin ligands expressed by neutrophils, monocytes, eosinophils, memory-effector T-like lymphocytes, natural killer cells or cancer cells. This interaction is associated with metastatic dissemination. However, the initial interaction between selectins and cancer cells are not sufficient to confer metastasis. As for leukocytes during inflammation, cancer cells bound to E-selectin are released into the circulation unless secondary adhesion mechanisms are activated. In some cases, cancer cells can interact with platelets and fibrinogen to form clots that further facilitate adhesion and spreading of the cancer cells to the endothelium of pulmonary vessels.
The extravasation of circulating tumor cells in the host organ requires successive adhesive interactions between endothelial cells and their ligands or counter-receptors present on the cancer cells. Thus, the specificity of binding between E-selectin and its ligands determine the organ selectivity in cancer metastasis.
Typically, the cancer cell/endothelial cell interactions imply first a selectin-mediated initial attachment and rolling of the circulating cancer cells on the endothelium. The rolling cancer cells then become activated by locally released chemokines present at the surface of endothelial cells. This triggers the activation of integrins from the cancer cells allowing their firmer adhesion to members of the Ig-CAM family such as ICAM, initiating the transendothelial migration and extravasation processes. The culture supernatants of cancer cells can trigger the expression of E-selectin by endothelial cells suggesting that cancer cells may release by themselves cytokines such as TNF-α, IL-1β or INF-γ that will directly activate endothelial cells to express E-selectin, P-selectin, ICAM-2 or VCAM.
Adhesion of colon cancer cells to endothelial cells expressing E-selectin induces a reverse signaling in the cancer cells that increases their motile potential, and a forward signaling in the endothelial cells that increases interendothelial permeability and enables extravasation. For example, adhesion of colon carcinoma cells to endothelial cells involves the binding of E- selectin on endothelial cells to death receptor-3 (DR3) on cancer cells. This interaction induces the reverse activation of p38 and ERK MAP kinases in cancer cells, which increases their motile and survival potentials. Reciprocally, the interaction between DR3 and E-selectin triggers the forward activation of the same MAP kinase pathways in endothelial cells. This results in myosin-light chain (MLC)-mediated cell retraction and in dissociation of the VE-cadherin/β-catenin complex and thereby destruction of adherens junctions leading to increased endothelial permeability and extravasation of cancer cells.
The process of E-selectin and endothelial adhesion receptor-mediated metastasis may be local. In particular, increased hepatic local metastasis of B16F1 melanoma cells is observed following exogenous IL-1a administration, which results from an increased vascular adhesion receptor expression, including E-selectin, VCAM-1 and ICAM-1, and tumor cell arrest in terminal portal venules.
Critical illness polyneuromyopathy
In cases of elevated blood glucose levels, such as in sepsis, E-selectin expression is higher than normal, resulting in greater microvascular permeability. The greater permeability leads to edema (swelling) of the skeletal endothelium (blood vessel linings), resulting in skeletal muscle ischemia (restricted blood supply) and eventually necrosis (cell death). This underlying pathology is the cause of the symptomatic disease critical illness polyneuromyopathy (CIPNM). Traditional Chinese herbal medicines, like berberine downregulate E-selectin.
Pathogen attachment
Study shows the adherence of porphyromonas gingivalis to human umbilical vein endothelial cells increases with the induction of E-selectin expression by TNF-α. An antibody to E-selectin and sialyl LewisX suppressed P. gingivalis adherence to stimulated HUVECs. P. gingivalis mutants lacking OmpA-like proteins Pgm6/7 had reduced adherence to stimulated HUVECs, but fimbriae-deficient mutants were not affected. E-selecin-mediated P. gingivalis adherence activated endothelial exocytosis. These results suggest that the interaction between host E-selectin and pathogen Pgm6/7 mediates P. gingivalis adherence to endothelial cells and may trigger vascular inflammation.
Acute coronary syndrome
The immunohistochemical expressions of E-selectin and PECAM-1 were significantly increased at intima in vulnerable plaques of acute coronary syndrome (ACS) group, especially in neovascular endothelial cells, and positively correlated with inflammatory cell density, suggesting that PECAM-1 and E-selectin might play an important role in inflammatory reaction and development of vulnerable plaque. E-selectin Ser128Arg polymorphism is associated with ACS, and it might be a risk factor for ACS.
Nicotine-mediated induction
Smoking is highly correlated with enhanced likelihood of atherosclerosis by inducing endothelial dysfunction. In endothelial cells, various cell-adhesion molecules including E-selectin, are shown to be upregulated upon exposure to nicotine, the addictive component of tobacco smoke. Nicotine-stimulated adhesion of monocytes to endothelial cells is dependent on the activation of α7-nAChRs, β-Arr1 and cSrc regulated increase in E2F1-mediated transcription of E-selectin gene. Therefore, agents such as RRD-251 that can target activity of E2F1 may have potential therapeutic benefit against cigarette smoke induced atherosclerosis.
Cerebral aneurysm
It's also found that E-selectin expression increased in human ruptured cerebral aneurysm tissues. E-selectin might be an important factor involved in the process of cerebral aneurysm formation and rupture, by promoting inflammation and weakening cerebral artery walls.
As a biomarker
E-selectin is also an emerging biomarker for the metastatic potential of some cancers including colorectal cancer and recurrences.