
Entrez 672 | Ensembl ENSG00000012048 | |
 | ||
Aliases BRCA1, breast cancer 1, early onset, BRCAI, BRCC1, BROVCA1, IRIS, PNCA4, PPP1R53, PSCP, RNF53, FANCS, breast cancer 1, DNA repair associated External IDs OMIM: 113705 MGI: 104537 HomoloGene: 5276 GeneCards: BRCA1 | ||
BRCA1 and BRCA1 (/ˌbrækəˈwʌn/) are a human gene and its protein product, respectively. The official symbol (BRCA1, italic for the gene, nonitalic for the protein) and the official name (breast cancer 1) are maintained by the HGNC. Orthologs, styled Brca1 and Brca1, are common in other mammal species. BRCA1 is a human tumor suppressor gene (to be specific, a caretaker gene), found in all humans; its protein, also called by the synonym breast cancer type 1 susceptibility protein, is responsible for repairing DNA.
Contents
- Discovery
- Gene location
- Protein structure
- Zinc ring finger domain
- Serine cluster domain
- BRCT domains
- Function and mechanism
- Transcription
- Mutations and cancer risk
- Low expression of BRCA1 in breast and ovarian cancers
- Mutation of BRCA1 in breast and ovarian cancer
- BRCA1 promoter hypermethylation in breast and ovarian cancer
- MicroRNA repression of BRCA1 in breast cancers
- MicroRNA repression of BRCA1 in ovarian cancers
- Deficiency of BRCA1 expression is likely tumorigenic
- Germ line mutations and founder effect
- Female fertility
- Cancer chemotherapy
- Patents enforcement litigation and controversy
- Interactions
- References
BRCA1 and BRCA2 are normally expressed in the cells of breast and other tissue, where they help repair damaged DNA, or destroy cells if DNA cannot be repaired. They are involved in the repair of chromosomal damage with an important role in the error-free repair of DNA double-strand breaks. If BRCA1 or BRCA2 itself is damaged by a BRCA mutation, damaged DNA is not repaired properly, and this increases the risk for breast cancer. Thus, although the terms "breast cancer susceptibility gene" and "breast cancer susceptibility protein" describe a proto-oncogene (a normal gene that could become an oncogene due to mutations or increased expression), BRCA1 and BRCA2 are normal; it is their mutation that is abnormal. Proto-oncogenes code for proteins that help to regulate cell growth and differentiation.
BRCA1 combines with other tumor suppressors, DNA damage sensors and signal transducers to form a large multi-subunit protein complex known as the BRCA1-associated genome surveillance complex (BASC). The BRCA1 protein associates with RNA polymerase II, and through the C-terminal domain, also interacts with histone deacetylase complexes. Thus, this protein plays a role in transcription, DNA repair of double-strand breaks ubiquitination, transcriptional regulation as well as other functions.
Methods to test for the likelihood of a patient with mutations in BRCA1 and BRCA2 developing cancer were covered by patents owned or controlled by Myriad Genetics. Myriad's business model of offering the diagnostic test exclusively led from Myriad being a startup in 1994 to being a publicly traded company with 1200 employees and about $500M in annual revenue in 2012; it also led to controversy over high prices and the inability to obtain second opinions from other diagnostic labs, which in turn led to the landmark Association for Molecular Pathology v. Myriad Genetics lawsuit.
Discovery
The first evidence for the existence of a gene encoding for a DNA repair enzyme involved in breast cancer susceptibility was provided by Mary-Claire King's laboratory at UC Berkeley in 1990. Four years later, after an international race to find it, the gene was cloned in 1994 by scientists at University of Utah, National Institute of Environmental Health Sciences (NIEHS) and Myriad Genetics.
Gene location
The human BRCA1 gene is located on the long (q) arm of chromosome 17 at region 2 band 1, from base pair 41,196,312 to base pair 41,277,500 (Build GRCh37/hg19) (map). BRCA1 orthologs have been identified in most mammals for which complete genome data are available.
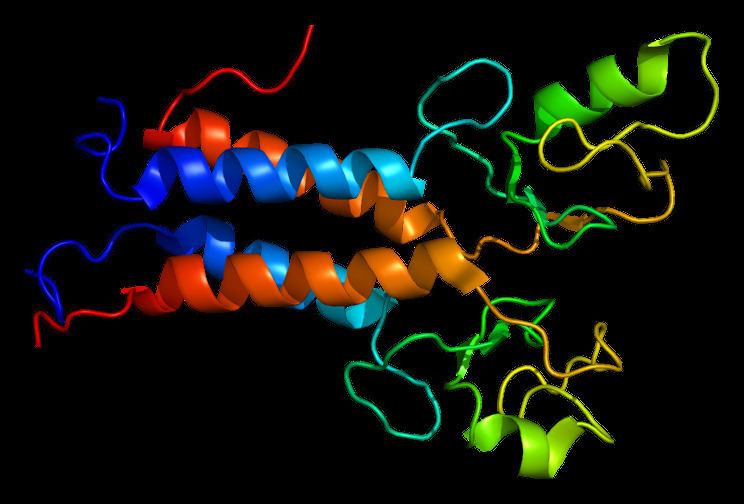
Protein structure
The BRCA1 protein contains the following domains:
This protein also contains nuclear localization signal and nuclear export signal motifs.
The human BRCA1 protein consists of four major protein domains; the Znf C3HC4- RING domain, the BRCA1 serine domain and two BRCT domains. These domains encode approximately 27% of BRCA1 protein. There are six known isoforms of BRCA1, with isoforms 1 and 2 comprising 1863 amino acids each.
Zinc ring finger domain
The RING motif, a Zn finger found in eukaryotic peptides, is 40–60 amino acids long and consists of eight conserved metal-binding residues, two quartets of cysteine or histidine residues that coordinate two zinc atoms. This motif contains a short anti-parallel beta-sheet, two zinc-binding loops and a central alpha helix in a small domain. This RING domain interacts with associated proteins, including BARD1, which also contains a RING motif, to form a heterodimer. The BRCA1 RING motif is flanked by alpha helices formed by residues 8–22 and 81–96 of the BRCA1 protein. It interacts with a homologous region in BARD1 also consisting of a RING finger flanked by two alpha-helices formed from residues 36–48 and 101–116. These four helices combine to form a heterodimerization interface and stabilise the BRCA1-BARD1 heterodimer complex. Additional stabilisation is achieved by interactions between adjacent residues in the flanking region and hydrophobic interactions. The BARD1/BRCA1 interaction is disrupted by tumorigenic amino acid substitutions in BRCA1, implying that the formation of a stable complex between these proteins may be an essential aspect of BRCA1 tumor suppression.
The ring domain is an important element of ubiquitin E3 ligases, which catalyse protein ubiquitination. Ubiquitin is a small regulatory protein found in all tissues that directs proteins to compartments within the cell. BRCA1 polypeptides, in particular Lys-48-linked polyubiquitin chains are dispersed throughout the resting cell nucleus, but at the start of DNA replication they gather in restrained groups that also contain BRCA2 and BARD1. BARD1 is thought to be involved in the recognition and binding of protein targets for ubiquitination. It attaches to proteins and labels them for destruction. Ubiquitination occurs via the BRCA1 fusion protein and is abolished by zinc chelation. The enzyme activity of the fusion protein is dependent on the proper folding of the ring domain.
Serine cluster domain
BRCA1 serine cluster domain (SCD) spans amino acids 1280–1524. A portion of the domain is located in exons 11–13. High rates of mutation occur in exons 11–13. Reported phosphorylation sites of BRCA1 are concentrated in the SCD, where they are phosphorylated by ATM/ATR kinases both in vitro and in vivo. ATM/ATR are kinases activated by DNA damage. Mutation of serine residues may affect localization of BRCA1 to sites of DNA damage and DNA damage response function.
BRCT domains
The dual repeat BRCT domain of the BRCA1 protein is an elongated structure approximately 70 Å long and 30–35 Å wide. The 85–95 amino acid domains in BRCT can be found as single modules or as multiple tandem repeats containing two domains. Both of these possibilities can occur in a single protein in a variety of different conformations. The C-terminal BRCT region of the BRCA1 protein is essential for repair of DNA, transcription regulation and tumor suppressor function. In BRCA1 the dual tandem repeat BRCT domains are arranged in a head-to-tail-fashion in the three-dimensional structure, burying 1600 Å of hydrophobic, solvent-accessible surface area in the interface. These all contribute to the tightly packed knob-in-hole structure that comprises the interface. These homologous domains interact to control cellular responses to DNA damage. It is, therefore, no surprise that a missense mutation at the interface of these two proteins can have devastating consequences on the cell cycle, resulting in protein dysfunction and a greater risk of developing cancer. The link that joins these two homologs also needs to be considered, since its poorly defined electron density alludes to a possible complex function; the ability to flex.
Function and mechanism
BRCA1 is part of a complex that repairs double-strand breaks in DNA. The strands of the DNA double helix are continuously breaking as they become damaged. Sometimes only one strand is broken, sometimes both strands are broken simultaneously. DNA cross-linking agents are an important source of chromosome/DNA damage. Double-strand breaks occur as intermediates after the crosslinks are removed, and indeed, biallelic mutations in BRCA1 have been identified to be responsible for Fanconi Anemia, Complementation Group S, a genetic disease associated with hypersensitivity to DNA crosslinking agents. BRCA1 is part of a protein complex that repairs DNA when both strands are broken. When this happens, it is difficult for the repair mechanism to "know" how to replace the correct DNA sequence, and there are multiple ways to attempt the repair. The double-strand repair mechanism in which BRCA1 participates is homologous recombination, where the repair proteins utilize a template of the identical homologous intact sequence from a sister chromatid, from a homologous chromosome, or from the same chromosome (depending on cell cycle phase). This DNA repair takes place with the DNA in the cell nucleus, wrapped around the histone. Several proteins, including BRCA1, arrive at the histone-DNA complex for this repair. Regulatory aspect to BRCA1 nuclear ⁄ non-nuclear distribution was first shown by the laboratory of Dr Rao in 1997
In the nucleus of many types of normal cells, the BRCA1 protein interacts with RAD51 during repair of DNA double-strand breaks. These breaks can be caused by natural radiation or other exposures, but also occur when chromosomes exchange genetic material (homologous recombination, e.g., "crossing over" during meiosis). The BRCA2 protein, which has a function similar to that of BRCA1, also interacts with the RAD51 protein. By influencing DNA damage repair, these three proteins play a role in maintaining the stability of the human genome.
BRCA1 is also involved in another type of DNA repair, termed mismatch repair. BRCA1 interacts with the DNA mismatch repair protein MSH2. MSH2, MSH6, PARP and some other proteins involved in single-strand repair are reported to be elevated in BRCA1-deficient mammary tumors.
A protein called valosin-containing protein (VCP, also known as p97) plays a role to recruit BRCA1 to the damaged DNA sites. After ionizing radiation, VCP is recruited to DNA lesions and cooperates with the ubiquitin ligase RNF8 to orchestrate assembly of signaling complexes for efficient DSB repair. BRCA1 interacts with VCP. BRCA1 also interacts with c-Myc, and other proteins that are critical to maintain genome stability.
BRCA1 directly binds to DNA, with higher affinity for branched DNA structures. This ability to bind to DNA contributes to its ability to inhibit the nuclease activity of the MRN complex as well as the nuclease activity of Mre11 alone. This may explain a role for BRCA1 to promote lower fidelity DNA repair by non-homologous end joining (NHEJ). BRCA1 also colocalizes with γ-H2AX (histone H2AX phosphorylated on serine-139) in DNA double-strand break repair foci, indicating it may play a role in recruiting repair factors.
Formaldehyde and acetaldehyde are common environmental sources of DNA cross links that often require repairs mediated by BRCA1 containing pathways.
This DNA repair function is essential; mice with loss-of-function mutations in both BRCA1 alleles are not viable, and as of 2015 only two adults were known to have loss-of-function mutations in both alleles; both had congenital or developmental issues, and both had cancer. One was presumed to have survived to adulthood because one of the BRCA1 mutations was hypomorphic.
Transcription
BRCA1 was shown to co-purify with the human RNA Polymerase II holoenzyme in HeLa extracts, implying it is a component of the holoenzyme. Later research, however, contradicted this assumption, instead showing that the predominant complex including BRCA1 in HeLa cells is a 2 megadalton complex containing SWI/SNF. SWI/SNF is a chromatin remodeling complex. Artificial tethering of BRCA1 to chromatin was shown to decondense heterochromatin, though the SWI/SNF interacting domain was not necessary for this role. BRCA1 interacts with the NELF-B (COBRA1) subunit of the NELF complex.
Mutations and cancer risk
Certain variations of the BRCA1 gene lead to an increased risk for breast cancer as part of a hereditary breast-ovarian cancer syndrome. Researchers have identified hundreds of mutations in the BRCA1 gene, many of which are associated with an increased risk of cancer. Women with an abnormal BRCA1 or BRCA2 gene have up to an 80% risk of developing breast cancer by age 90; increased risk of developing ovarian cancer is about 55% for women with BRCA1 mutations and about 25% for women with BRCA2 mutations.
These mutations can be changes in one or a small number of DNA base pairs (the building-blocks of DNA), and can be identified with PCR and DNA sequencing.
In some cases, large segments of DNA are rearranged. Those large segments, also called large rearrangements, can be a deletion or a duplication of one or several exons in the gene. Classical methods for mutation detection (sequencing) are unable to reveal these types of mutation. Other methods have been proposed: traditional quantitative PCR, Multiplex Ligation-dependent Probe Amplification (MLPA), and Quantitative Multiplex PCR of Short Fluorescent Fragments (QMPSF). Newer methods have also been recently proposed: heteroduplex analysis (HDA) by multi-capillary electrophoresis or also dedicated oligonucleotides array based on comparative genomic hybridization (array-CGH).
Some results suggest that hypermethylation of the BRCA1 promoter, which has been reported in some cancers, could be considered as an inactivating mechanism for BRCA1 expression.
A mutated BRCA1 gene usually makes a protein that does not function properly. Researchers believe that the defective BRCA1 protein is unable to help fix DNA damage leading to mutations in other genes. These mutations can accumulate and may allow cells to grow and divide uncontrollably to form a tumor. Thus, BRCA1 inactivating mutations lead to a predisposition for cancer.
BRCA1 mRNA 3' UTR can be bound by an miRNA, Mir-17 microRNA. It has been suggested that variations in this miRNA along with Mir-30 microRNA could confer susceptibility to breast cancer.
In addition to breast cancer, mutations in the BRCA1 gene also increase the risk of ovarian and prostate cancers. Moreover, precancerous lesions (dysplasia) within the Fallopian tube have been linked to BRCA1 gene mutations. Pathogenic mutations anywhere in a model pathway containing BRCA1 and BRCA2 greatly increase risks for a subset of leukemias and lymphomas.
Women having inherited a defective BRCA1 or BRCA2 gene have risks for breast and ovarian cancer that are so high and seem so selective that many mutation carriers choose to have prophylactic surgery. There has been much conjecture to explain such apparently striking tissue specificity. Major determinants of where BRCA1/2 hereditary cancers occur are related to tissue specificity of the cancer pathogen, the agent that causes chronic inflammation or the carcinogen. The target tissue may have receptors for the pathogen, may become selectively exposed to an inflammatory process or to a carcinogen. An innate genomic deficit in a tumor suppressor gene impairs normal responses and exacerbates the susceptibility to disease in organ targets. This theory also fits data for several tumor suppressors beyond BRCA1 or BRCA2. A major advantage of this model is that it suggests there may be some options in addition to prophylactic surgery.
Low expression of BRCA1 in breast and ovarian cancers
BRCA1 expression is reduced or undetectable in the majority of high grade, ductal breast cancers. It has long been noted that loss of BRCA1 activity, either by germ-line mutations or by down-regulation of gene expression, leads to tumor formation in specific target tissues. In particular, decreased BRCA1 expression contributes to both sporadic and inherited breast tumor progression. Reduced expression of BRCA1 is tumorigenic because it plays an important role in the repair of DNA damages, especially double-strand breaks, by the potentially error-free pathway of homologous recombination. Since cells that lack the BRCA1 protein tend to repair DNA damages by alternative more error-prone mechanisms, the reduction or silencing of this protein generates mutations and gross chromosomal rearrangements that can lead to progression to breast cancer.
Similarly, BRCA1 expression is low in the majority (55%) of sporadic epithelial ovarian cancers (EOCs) where EOCs are the most common type of ovarian cancer, representing approximately 90% of ovarian cancers. In serous ovarian carcinomas, a sub-category constituting about 2/3 of EOCs, low BRCA1 expression occurs in more than 50% of cases. Bowtell reviewed the literature indicating that deficient homologous recombination repair caused by BRCA1 deficiency is tumorigenic. In particular this deficiency initiates a cascade of molecular events that sculpt the evolution of high-grade serous ovarian cancer and dictate its response to therapy. Especially noted was that BRCA1 deficiency could be the cause of tumorigenesis whether due to BRCA1 mutation or any other event that causes a deficiency of BRCA1 expression.
Mutation of BRCA1 in breast and ovarian cancer
Only about 3%-8% of all women with breast cancer carry a mutation in BRCA1 or BRCA2. Similarly, BRCA1 mutations are only seen in about 18% of ovarian cancers (13% germline mutations and 5% somatic mutations).
Thus, while BRCA1 expression is low in the majority of these cancers, BRCA1 mutation is not a major cause of reduced expression.
BRCA1 promoter hypermethylation in breast and ovarian cancer
BRCA1 promoter hypermethylation was present in only 13% of unselected primary breast carcinomas. Similarly, BRCA1 promoter hypermethylation was present in only 5% to 15% of EOC cases.
Thus, while BRCA1 expression is low in these cancers, BRCA1 promoter methylation is only a minor cause of reduced expression.
MicroRNA repression of BRCA1 in breast cancers
There are a number of specific microRNAs, when overexpressed, that directly reduce expression of specific DNA repair proteins (see MicroRNA section DNA repair and cancer) In the case of breast cancer, microRNA-182 (miR-182) specifically targets BRCA1. Breast cancers can be classified based on receptor status or histology, with triple-negative breast cancer (15% - 25% of breast cancers), HER2+ (15% - 30% of breast cancers), ER+/PR+ (about 70% of breast cancers), and Invasive lobular carcinoma (about 5% - 10% of invasive breast cancer). All four types of breast cancer were found to have an average of about 100-fold increase in miR-182, compared to normal breast tissue. In breast cancer cell lines, there is an inverse correlation of BRCA1 protein levels with miR-182 expression. Thus it appears that much of the reduction or absence of BRCA1 in high grade ductal breast cancers may be due to over-expressed miR-182.
In addition to miR-182, a pair of almost identical microRNAs, miR-146a and miR-146b-5p, also repress BRCA1 expression. These two microRNAs are over-expressed in triple-negative tumors and their over-expression results in BRCA1 inactivation. Thus, miR-146a and/or miR-146b-5p may also contribute to reduced expression of BRCA1 in these triple-negative breast cancers.
MicroRNA repression of BRCA1 in ovarian cancers
In both serous tubal intraepithelial carcinoma (the precursor lesion to high grade serous ovarian carcinoma (HG-SOC)), and in HG-SOC itself, miR-182 is overexpressed in about 70% of cases. In cells with over-expressed miR-182, BRCA1 remained low, even after exposure to ionizing radiation (which normally raises BRCA1 expression). Thus much of the reduced or absent BRCA1 in HG-SOC may be due to over-expressed miR-182.
Another microRNA known to reduce expression of BRCA1 in ovarian cancer cells is miR-9. Among 58 tumors from patients with stage IIIC or stage IV serous ovarian cancers (HG-SOG), an inverse correlation was found between expressions of miR-9 and BRCA1, so that increased miR-9 may also contribute to reduced expression of BRCA1 in these ovarian cancers.
Deficiency of BRCA1 expression is likely tumorigenic
DNA damage appears to be the primary underlying cause of cancer, and deficiencies in DNA repair appears to underlie many forms of cancer. If DNA repair is deficient, DNA damage tends to accumulate. Such excess DNA damage may increase mutational errors during DNA replication due to error-prone translesion synthesis. Excess DNA damage may also increase epigenetic alterations due to errors during DNA repair. Such mutations and epigenetic alterations may give rise to cancer. The frequent microRNA-induced deficiency of BRCA1 in breast and ovarian cancers likely contribute to the progression of those cancers.
Germ line mutations and founder effect
All germ-line BRCA1 mutations identified to date have been inherited, suggesting the possibility of a large “founder” effect in which a certain mutation is common to a well-defined population group and can, in theory, be traced back to a common ancestor. Given the complexity of mutation screening for BRCA1, these common mutations may simplify the methods required for mutation screening in certain populations. Analysis of mutations that occur with high frequency also permits the study of their clinical expression. Examples of manifestations of a founder effect are seen among Ashkenazi Jews. Three mutations in BRCA1 have been reported to account for the majority of Ashkenazi Jewish patients with inherited BRCA1-related breast and/or ovarian cancer: 185delAG, 188del11 and 5382insC in the BRCA1 gene. In fact, it has been shown that if a Jewish woman does not carry a BRCA1 185delAG, BRCA1 5382insC founder mutation, it is highly unlikely that a different BRCA1 mutation will be found. Additional examples of founder mutations in BRCA1 are given in Table 1 (mainly derived from ).
Female fertility
As women age, their reproductive performance declines, leading to menopause. This decline is tied to a reduction in the number of ovarian follicles. Although about 1 million oocytes are present at birth in the human ovary, only about 500 (about 0.05%) of these ovulate, and the rest are wasted. The decline in ovarian reserve appears to occur at a constantly increasing rate with age, and leads to nearly complete exhaustion of the reserve by about age 52. As ovarian reserve and fertility decline with age, there is also a parallel increase in pregnancy failure and meiotic errors, resulting in chromosomally abnormal conceptions.
Women with a germ-line BRCA1 mutation appear to have a diminished oocyte reserve and decreased fertility compared to normally aging women. Furthermore, women with an inherited BRCA1 mutation undergo menopause prematurely. Since BRCA1 is a key DNA repair protein, these findings suggest that naturally occurring DNA damages in oocytes are repaired less efficiently in women with a BRCA1 defect, and that this repair inefficiency leads to early reproductive failure.
As noted above, the BRCA1 protein plays a key role in homologous recombinational repair. This is the only known cellular process that can accurately repair DNA double-strand breaks. DNA double-strand breaks accumulate with age in humans and mice in primordial follicles. Primordial follicles contain oocytes that are at an intermediate (prophase I) stage of meiosis. Meiosis is the general process in eukaryotic organisms by which germ cells are formed, and it is likely an adaptation for removing DNA damages, especially double-strand breaks, from germ line DNA. (Also see article Meiosis). Homologous recombinational repair employing BRCA1 is especially promoted during meiosis. It was found that expression of 4 key genes necessary for homologous recombinational repair of DNA double-strand breaks (BRCA1, MRE11, RAD51 and ATM) decline with age in the oocytes of humans and mice, leading to the hypothesis that DNA double-strand break repair is necessary for the maintenance of oocyte reserve and that a decline in efficiency of repair with age plays a role in ovarian aging.
Cancer chemotherapy
Non-small cell lung cancer (NSCLC) is the leading cause of cancer deaths worldwide. At diagnosis, almost 70% of persons with NSCLC have locally advanced or metastatic disease. Persons with NSCLC are often treated with therapeutic platinum compounds (e.g. cisplatin, carboplatin or oxaliplatin) that cause inter-strand cross-links in DNA. Among individuals with NSCLC, low expression of BRCA1 in the primary tumor correlated with improved survival after platinum-containing chemotherapy. This correlation implies that low BRCA1 in the cancer, and the consequent low level of DNA repair, causes vulnerability of the cancer to treatment by the DNA cross-linking agents. High BRCA1 may protect cancer cells by acting in a pathway that removes the damages in DNA introduced by the platinum drugs. Thus the level of BRCA1 expression is a potentially important tool for tailoring chemotherapy in lung cancer management.
Level of BRCA1 expression is also relevant to ovarian cancer treatment. Patients having sporadic ovarian cancer who were treated with platinum drugs had longer median survival times if their BRCA1 expression was low compared to patients with higher BRCA1 expression (46 compared to 33 months).
Patents, enforcement, litigation, and controversy
A patent application for the isolated BRCA1 gene and cancer promoting mutations discussed above, as well as methods to diagnose the likelihood of getting breast cancer, was filed by the University of Utah, National Institute of Environmental Health Sciences (NIEHS) and Myriad Genetics in 1994; over the next year, Myriad, (in collaboration with investigators at Endo Recherche, Inc., HSC Research & Development Limited Partnership, and University of Pennsylvania), isolated and sequenced the BRCA2 gene and identified key mutations, and the first BRCA2 patent was filed in the U.S. by Myriad and other institutions in 1995. Myriad is the exclusive licensee of these patents and has enforced them in the US against clinical diagnostic labs. This business model led from Myriad being a startup in 1994 to being a publicly traded company with 1200 employees and about $500M in annual revenue in 2012; it also led to controversy over high prices and the inability to get second opinions from other diagnostic labs, which in turn led to the landmark Association for Molecular Pathology v. Myriad Genetics lawsuit. The patents began to expire in 2014.
According to an article published in the journal, Genetic Medicine, in 2010, "The patent story outside the United States is more complicated.... For example, patents have been obtained but the patents are being ignored by provincial health systems in Canada. In Australia and the UK, Myriad’s licensee permitted use by health systems, but announced a change of plans in August 2008. Only a single mutation has been patented in Myriad’s lone European-wide patent, although some patents remain under review of an opposition proceeding. In effect, the United States is the only jurisdiction where Myriad’s strong patent position has conferred sole-provider status." Peter Meldrum, CEO of Myriad Genetics, has acknowledged that Myriad has "other competitive advantages that may make such [patent] enforcement unnecessary" in Europe.
Legal decisions surrounding the BRCA1 and BRCA2 patents will affect the field of genetic testing in general. A June 2013 article, in Association for Molecular Pathology v. Myriad Genetics (No. 12-398), quoted the US Supreme Court's unanimous ruling that, "A naturally occurring DNA segment is a product of nature and not patent eligible merely because it has been isolated," invalidating Myriad's patents on the BRCA1 and BRCA2 genes. However, the Court also held that manipulation of a gene to create something not found in nature could still be eligible for patent protection. The Federal Court of Australia came to the opposite conclusion, upholding the validity of an Australian Myriad Genetics patent over the BRCA1 gene in February 2013. The Federal Court also rejected an appeal in September 2014. Yvonne D’Arcy won her case against US-based biotech company Myriad Genetics in the High Court of Australia. In their unanimous decision on October 7, 2015 the "high court found that an isolated nucleic acid, coding for a BRCA1 protein, with specific variations from the norm that are indicative of susceptibility to breast cancer and ovarian cancer was not a 'patentable invention.'"
Interactions
BRCA1 has been shown to interact with the following proteins: