
Symbol DNA_mis_repair Pfam clan CL0329 PROSITE PDOC00057 | Pfam PF01119 InterPro IPR013507 SCOP 1bkn | |
 | ||
DNA mismatch repair is a system for recognizing and repairing erroneous insertion, deletion, and mis-incorporation of bases that can arise during DNA replication and recombination, as well as repairing some forms of DNA damage.
Contents
- Mismatch repair proteins
- MutS homologs
- MutL homologs
- MutH an endonuclease present in E coli and Salmonella
- PCNA sliding clamp
- Defects in mismatch repair
- Epigenetic defects in cancer
- Deficiency in field defects
- MMR deficiency in humans
- Lack of MMR and mutation frequency
- References
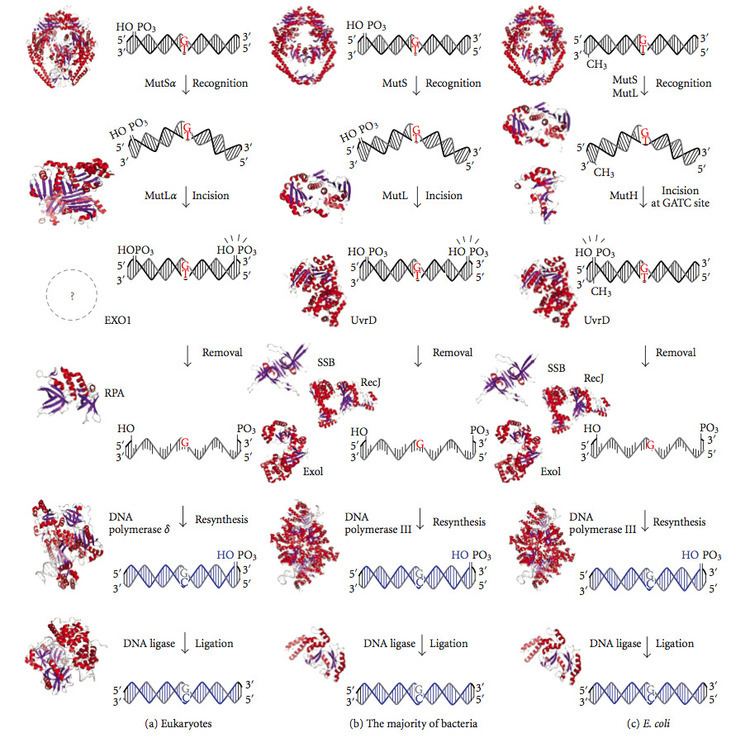
Mismatch repair is strand-specific. During DNA synthesis the newly synthesised (daughter) strand will commonly include errors. In order to begin repair, the mismatch repair machinery distinguishes the newly synthesised strand from the template (parental). In gram-negative bacteria, transient hemimethylation distinguishes the strands (the parental is methylated and daughter is not). However, in other prokaryotes and eukaryotes, the exact mechanism is not clear. It is suspected that, in eukaryotes, newly synthesized lagging-strand DNA transiently contains nicks (before being sealed by DNA ligase) and provides a signal that directs mismatch proofreading systems to the appropriate strand. This implies that these nicks must be present in the leading strand, and evidence for this has recently been found. Recent work has shown that nicks are sites for RFC-dependent loading of the replication sliding clamp PCNA, in an orientation-specific manner, such that one face of the donut-shape protein is juxtaposed toward the 3'-OH end at the nick. Oriented PCNA then directs the action of the MutLalpha endonuclease to one strand in the presence of a mismatch and MutSalpha or MutSbeta.
Any mutational event that disrupts the superhelical structure of DNA carries with it the potential to compromise the genetic stability of a cell. The fact that the damage detection and repair systems are as complex as the replication machinery itself highlights the importance evolution has attached to DNA fidelity.
Examples of mismatched bases include a G/T or A/C pairing (see DNA repair). Mismatches are commonly due to tautomerization of bases during DNA replication. The damage is repaired by recognition of the deformity caused by the mismatch, determining the template and non-template strand, and excising the wrongly incorporated base and replacing it with the correct nucleotide. The removal process involves more than just the mismatched nucleotide itself. A few or up to thousands of base pairs of the newly synthesized DNA strand can be removed.
Mismatch repair proteins
Mismatch repair is a highly conserved process from prokaryotes to eukaryotes. The first evidence for mismatch repair was obtained from S. pneumoniae (the hexA and hexB genes). Subsequent work on E. coli has identified a number of genes that, when mutationally inactivated, cause hypermutable strains. The gene products are, therefore, called the "Mut" proteins, and are the major active components of the mismatch repair system. Three of these proteins are essential in detecting the mismatch and directing repair machinery to it: MutS, MutH and MutL (MutS is a homologue of HexA and MutL of HexB).
MutS forms a dimer (MutS2) that recognises the mismatched base on the daughter strand and binds the mutated DNA. MutH binds at hemimethylated sites along the daughter DNA, but its action is latent, being activated only upon contact by a MutL dimer (MutL2), which binds the MutS-DNA complex and acts as a mediator between MutS2 and MutH, activating the latter. The DNA is looped out to search for the nearest d(GATC) methylation site to the mismatch, which could be up to 1 kb away. Upon activation by the MutS-DNA complex, MutH nicks the daughter strand near the hemimethylated site and recruits a UvrD helicase (DNA Helicase II) to separate the two strands with a specific 3' to 5' polarity. The entire MutSHL complex then slides along the DNA in the direction of the mismatch, liberating the strand to be excised as it goes. An exonuclease trails the complex and digests the ss-DNA tail. The exonuclease recruited is dependent on which side of the mismatch MutH incises the strand – 5' or 3'. If the nick made by MutH is on the 5' end of the mismatch, either RecJ or ExoVII (both 5' to 3' exonucleases) is used. If, however, the nick is on the 3' end of the mismatch, ExoI (a 3' to 5' enzyme) is used.
The entire process ends past the mismatch site - i.e., both the site itself and its surrounding nucleotides are fully excised. The single-strand gap created by the exonuclease can then be repaired by DNA Polymerase III (assisted by single-strand-binding protein), which uses the other strand as a template, and finally sealed by DNA ligase. DNA methylase then rapidly methylates the daughter strand.
MutS homologs
When bound, the MutS2 dimer bends the DNA helix and shields approximately 20 base pairs. It has weak ATPase activity, and binding of ATP leads to the formation of tertiary structures on the surface of the molecule. The crystal structure of MutS reveals that it is exceptionally asymmetric, and, while its active conformation is a dimer, only one of the two halves interacts with the mismatch site.
In eukaryotes, MutS homologs form two major heterodimers: Msh2/Msh6 (MutSα) and Msh2/Msh3 (MutSβ). The MutSα pathway is involved primarily in base substitution and small-loop mismatch repair. The MutSβ pathway is also involved in small-loop repair, in addition to large-loop (~10 nucleotide loops) repair. However, MutSβ does not repair base substitutions.
MutL homologs
MutL also has weak ATPase activity (it uses ATP for purposes of movement). It forms a complex with MutS and MutH, increasing the MutS footprint on the DNA.
However, the processivity (the distance the enzyme can move along the DNA before dissociating) of UvrD is only ~40–50 bp. Because the distance between the nick created by MutH and the mismatch can average ~600 bp, if there is not another UvrD loaded the unwound section is then free to re-anneal to its complementary strand, forcing the process to start over. However, when assisted by MutL, the rate of UvrD loading is greatly increased. While the processivity (and ATP utilisation) of the individual UvrD molecules remains the same, the total effect on the DNA is boosted considerably; the DNA has no chance to re-anneal, as each UvrD unwinds 40-50 bp of DNA, dissociates, and then is immediately replaced by another UvrD, repeating the process. This exposes large sections of DNA to exonuclease digestion, allowing for quick excision (and later replacement) of the incorrect DNA.
Eukaryotes have MutL homologs designated Mlh1 and Pms1. They form a heterodimer that mimics MutL in E. coli. The human homologue of prokaryotic MutL has three forms designated as MutLα, MutLβ, and MutLγ. The MutLα complex is made of two subunits MLH1 and PMS2, the MutLβ heterodimer is made of MLH1 and PMS1, whereas MutLγ is made of MLH1 and MLH3. MutLα acts as the matchmaker or facilitator, coordinating events in mismatch repair. It has recently been shown to be a DNA endonuclease that introduces strand breaks in DNA upon activation by mismatch and other required proteins, MutSa and PCNA. These strand interruptions serve as entry points for an exonuclease activity that removes mismatched DNA. Roles played by MutLβ and MutLγ in mismatch repair are less-understood.
MutH: an endonuclease present in E. coli and Salmonella
MutH is a very weak endonuclease that is activated once bound to MutL (which itself is bound to MutS). It nicks unmethylated DNA and the unmethylated strand of hemimethylated DNA but does not nick fully methylated DNA. Experiments have shown that mismatch repair is random if neither strand is methylated. These behaviours led to the proposal that MutH determines which strand contains the mismatch. MutH has no eukaryotic homolog. Its endonuclease function is taken up by MutL homologs, which have some specialized 5'-3' exonuclease activity. The strand bias for removing mismatches from the newly synthesized daughter strand in eukaryotes may be provided by the free 3' ends of Okazaki fragments in the new strand created during replication.
PCNA β-sliding clamp
PCNA and the β-sliding clamp associate with MutSα/β and MutS, respectively. Although initial reports suggested that the PCNA-MutSα complex may enhance mismatch recognition, it has been recently demonstrated that there is no apparent change in affinity of MutSα for a mismatch in the presence or absence of PCNA. Furthermore, mutants of MutSα that are unable to interact with PCNA in vitro exhibit the capacity to carry out mismatch recognition and mismatch excision to near wild type levels. Such mutants are defective in the repair reaction directed by a 5' strand break, suggesting for the first time MutSα function in a post-excision step of the reaction.
Defects in mismatch repair
Mutations in the human homologues of the Mut proteins affect genomic stability, which can result in microsatellite instability (MI). MI is implicated in most human cancers. To be specific, the overwhelming majority of hereditary nonpolyposis colorectal cancers (HNPCC) are attributed to mutations in the genes encoding the MutS and MutL homologues MSH2 and MLH1 respectively, which allows them to be classified as tumour suppressor genes. A subtype of HNPCC is known as Muir-Torre Syndrome (MTS), which is associated with skin tumors.
Mismatch repair cancer syndrome (also called mismatch repair deficiency or Turcot syndrome) typically combines familial adenomatous polyposis with brain tumors.
Epigenetic defects in cancer
Only a minority of sporadic cancers with a DNA repair deficiency have a mutation in a DNA repair gene. However, a majority of sporadic cancers with a DNA repair deficiency do have one or more epigenetic alterations that reduce or silence DNA repair gene expression. About 13% of colorectal cancers are deficient in DNA mismatch repair, with 9.8% due to loss of MLH1 and smaller percentages due to losses of MSH2 (1.4%), MSH6 (0.5%) and PMS2 (1.5%). For 65 out of 66 cases of sporadic cancer in which MLH1 was deficient, the deficiency was due to methylation of the promoter region of MLH1.
Other cancers have higher frequencies of loss of MMR due to loss of MLH1 (see table, below). As indicated in the table, defects in MMR that were due to loss of MLH1 expression were evaluated, and it was found that deficiencies of MLH1 were largely a result of methylation of the promoter region of the MLH1 gene.
Another epigenetic mechanism by which MLH1 and MSH2 expression is reduced is over-expression of miR-155. MiR-155 targets MLH1 and MSH2. In human colorectal cancer an inverse correlation between the expression of miR-155 and the expression of MLH1 or MSH2 proteins was found.
Deficiency in field defects
A field defect is an area or "field" of epithelium that has been preconditioned by epigenetic changes and/or mutations so as to predispose it towards development of cancer. As pointed out by Rubin, "The vast majority of studies in cancer research has been done on well-defined tumors in vivo, or on discrete neoplastic foci in vitro. Yet there is evidence that more than 80% of the somatic mutations found in mutator phenotype human colorectal tumors occur before the onset of terminal clonal expansion." Similarly, Vogelstein et al. point out that more than half of somatic mutations identified in tumors occurred in a pre-neoplastic phase (in a field defect), during growth of apparently normal cells.
In the Table above, MLH1 deficiencies were noted in the field defects (histologically normal tissues) surrounding most of the cancers. If MLH1 is epigenetically reduced or silenced, it would not likely confer a selective advantage upon a stem cell. However, reduced or absent expression of MLH1 would cause increased rates of mutation, and one or more of the mutated genes may provide the cell with a selective advantage. The expression-deficient MLH1 gene could then be carried along as a selectively neutral or only slightly deleterious passenger (hitch-hiker) gene when the mutated stem cell generates an expanded clone. The continued presence of a clone with an epigenetically repressed MLH1 would continue to generate further mutations, some of which could produce a tumor.
MMR deficiency in humans
In humans, seven DNA mismatch repair (MMR) proteins (MLH1, MLH3, MSH2, MSH3, MSH6, PMS1 and PMS2) work coordinately in sequential steps to initiate repair of DNA mismatches. In addition, there are Exo1-dependent and Exo1-independent MMR subpathways.
Other gene products involved in mismatch repair (subsequent to initiation by MMR genes) in humans include DNA polymerase delta, PCNA, RPA, HMGB1, RFC and DNA ligase I, plus histone and chromatin modifying factors.
Lack of MMR and mutation frequency
Recognizing mismatches and repairing them is important for cells because failure to do so results in microsatellite instability (MSI) and an elevated spontaneous mutation rate (mutator phenotype). Among 20 cancers evaluated, mismatch repair deficient microsatellite instable (MSI) colon cancer had the second highest frequency of mutations (after melanoma).
However, lack of MMR often occurs in coordination with loss of other DNA repair genes. In one example, involving MLH1 and MLH3, Jiang et al. conducted a study where they evaluated the mRNA expression of 27 DNA repair genes in 40 astrocytomas compared to normal brain tissues from non-astrocytoma individuals. Among the 27 DNA repair genes evaluated, 13 DNA repair genes, MLH1, MLH3, MGMT, NTHL1, OGG1, SMUG1, ERCC1, ERCC2, ERCC3, ERCC4, RAD50, XRCC4 and XRCC5 were all significantly down-regulated in all three grades (II, III and IV) of astrocytomas. The repression of these 13 genes in lower grade as well as in higher grade astrocytomas suggested that they may be important in early as well as in later stages of astrocytoma. In another example, Kitajima et al. found that immunoreactivity for MLH1 and MGMT expression was closely correlated in 135 specimens of gastric cancer and loss of MLH1 and MGMT appeared to be synchronously accelerated during tumor progression.
Deficient expression of multiple DNA repair genes is often found in cancers, and may contribute to the thousands of mutations usually found in cancers (see Mutation frequencies in cancers).