
Formula C4H6O4 IUPAC ID Butanedioic acid Boiling point 235 °C Soluble in Water | Molar mass 118.09 g/mol Melting point 184 °C Density 1.56 g/cm³ | |
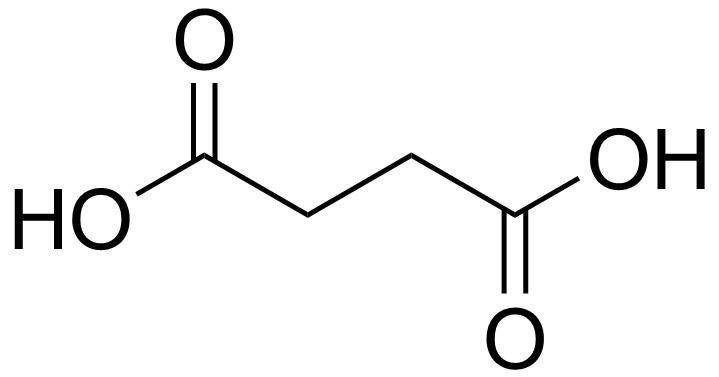 | ||
Succinic acid (/səkˈsɪnᵻk/) is a dicarboxylic acid with the chemical formula (CH2)2(CO2H)2. The name derives from Latin succinum, meaning amber. In living organisms, succinic acid takes the form of an anion, succinate, which has multiple biological roles both as a metabolic intermediate and as a signaling molecule reflecting the cellular metabolic state. Succinate is generated in mitochondria via the tricarboxylic acid cycle (TCA), an ancient energy-yielding process shared by all organisms. Succinate can exit the mitochondrial matrix and function in the cytoplasm as well as the extracellular space, changing gene expression patterns, modulating epigenetic landscape or demonstrating hormone-like signaling. As such, succinate links cellular metabolism to the regulation of cellular function. Dysregulation of succinate synthesis and degradation can lead to pathological conditions, such as malignant transformation, inflammation and tissue injury.
Contents
- Bioamber commericalizes bio based succinic acid chemistry inspired by nature
- Physical properties
- Chemical reactions
- Commercial production
- Applications
- Precursor to polymers resins and solvents
- Food and dietary supplement
- Tricarboxylic acid TCA cycle
- Reductive branch of the TCA cycle
- Glyoxylate cycle
- GABA shunt
- Metabolic intermediate
- ROS production
- Additional biologic functions
- Succinate transporters
- Extracellular signaling
- Effect on adipocytes
- Effect on the liver and retina
- Effect on the heart
- Effect on immune cells
- Effect on platelets
- Effect on the kidneys
- Intracellular signaling
- Epigenetic effects
- Gene regulation
- Inflammation
- Tumorigenesis
- Ischemia reperfusion injury
- References
Bioamber commericalizes bio based succinic acid chemistry inspired by nature
Physical properties
Succinic acid is a white, odorless solid with a highly acidic taste. In an aqueous solution, succinic acid readily ionizes to form its conjugate base, succinate (/ˈsʌksᵻneɪt/). As a diprotic acid, succinic acid undergoes two successive deprotonation reactions:
(CH2)2(CO2H)2 → (CH2)2(CO2H)(CO2)− + H+(CH2)2(CO2H)(CO2)− → (CH2)2(CO2)22− + H+
The pKa of these processes are 4.3 and 5.6, respectively. Both anions are colorless and can be isolated as the salts, e.g., Na(CH2)2(CO2H)(CO2) and Na2(CH2)2(CO2)22−. In living organisms, primarily succinate, not succinic acid, is found.
As a radical group it is called a succinyl (/ˈsʌksᵻnəl/) group.
Like most simple mono- and dicarboxylic acids, it is not harmful but can be an irritant to skin and eyes.
Chemical reactions

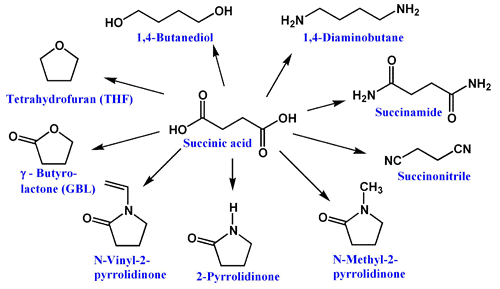
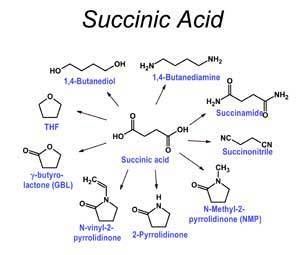
Succinic acid can be oxidized to fumaric acid or be converted to diesters, such as diethylsuccinate (CH2CO2CH2CH3)2. This diethyl ester is a substrate in the Stobbe condensation. Dehydration of succinic acid gives succinic anhydride. Succinate can be used to derive 1,4-butanediol, maleic anhydride, succinimide, 2-pyrrolidinone and tetrahydrofuran.
Commercial production
Historically, succinic acid was obtained from amber by distillation and has thus been known as spirit of amber. Today, succinic acid is generated for human use synthetically or converted from biomass via fermentation. Common industrial routes of synthesis include partial hydrogenation of maleic acid, oxidation of 1,4-butanediol, and carbonylation of ethylene glycol. Succinate is also produced petrochemically from butane via maleic anhydride. Additionally, genetic engineering of bacteria, such as Escherichia coli or Saccharomyces cerevisiae, has recently allowed for the high-yielding, commercial production from fermentation of glucose. Global production is estimated at 16,000 to 30,000 tons a year, with an annual growth rate of 10%.
Applications
In 2004, succinate was placed on the US Department of Energy's list of top 12 platform chemicals from biomass.
Precursor to polymers, resins, and solvents
Succinic acid is a precursor to some polyesters and a component of some alkyd resins. A key derivative of succinic acid, 1,4-butanediol (BDO), has major industrial applications. The global market size for BDO was estimated at USD 4.72 billion in 2013. BDO is a precursor to the common organic solvent THF and a precursor to polybutylene terephthalate (PBT), an engineering-grade thermoplastic. The automotive and electronics industries heavily rely on PBT to produce connectors, insulators, wheel covers, gearshift knobs and reinforcing beams. Succinic acid also serves as the bases of certain biodegradable polymers, which are of interest in tissue engineering applications.
Food and dietary supplement
As a food additive and dietary supplement, succinic acid is generally recognized as safe by the U.S. Food and Drug Administration. Succinic acid is used primarily as an acidity regulator in the food and beverage industry. It is also available as a flavoring agent, contributing a somewhat sour and astringent component to umami taste. As an excipient in pharmaceutical products, it is also used to control acidity or as a counter ion. Drugs involving succinate include metoprolol succinate, sumatriptan succinate or solifenacin succinate.
Tricarboxylic acid (TCA) cycle
Succinate is a key intermediate in the tricarboxylic acid cycle, a primary metabolic pathway used to produce chemical energy in the presence of O2. Succinate is generated from succinyl-CoA by the enzyme succinyl-CoA synthetase in a GTP/ATP-producing step:
Succinyl-CoA + NDP + Pi → Succinate + CoA + NTP
Catalyzed by the enzyme succinate dehydrogenase (SDH), succinate is subsequently oxidized to fumarate:
Succinate + FAD → Fumarate + FADH2
SDH also participates in the mitochondrial electron transport chain, where it is known as respiratory Complex 2. This enzyme complex is a 4 subunit membrane-bound lipoprotein which couples the oxidation of succinate to the reduction of ubiquinone via the intermediate electron carriers FAD and three 2Fe-2S clusters. Succinate thus serves as a direct electron donor to the electron transport chain.
Click on genes, proteins and metabolites below to link to respective articles.
Reductive branch of the TCA cycle
Succinate can alternatively be formed by reverse activity of SDH. Under anaerobic conditions certain bacteria such as A. succinogenes, A. succiniciproducens and M. succiniciproducens, run the TCA cycle in reverse and convert glucose to succinate through the intermediates of oxaloacetate, malate and fumarate. This pathway is exploited in metabolic engineering to net generate succinate for human use. Additionally, succinic acid produced during the fermentation of sugar provides a combination of saltiness, bitterness and acidity to fermented alcohols.
Accumulation of fumarate can drive the reverse activity of SDH, thus enhancing succinate generation. Under pathological and physiological conditions, the malate-aspartate shuttle or the purine nucleotide shuttle can increase mitochondrial fumarate, which is then readily converted to succinate.
Glyoxylate cycle
Succinate is also a product of the glyoxylate cycle, which converts two two-carbon acetyl units into the four-carbon succinate. The glyoxylate cycle is utilized by many bacteria, plants and fungi and allows these organisms to subsist on acetate or acetyl CoA yielding compounds. The pathway avoids the decarboxylation steps of the TCA cycle via the enzyme isocitrate lyase which cleaves isocitrate into succinate and glyoxylate. Generated succinate is then available for either energy production or biosynthesis.
GABA shunt
Succinate is the re-entry point for the gamma-aminobutyric acid (GABA) shunt into the TCA cycle, a closed cycle which synthesizes and recycles GABA. The GABA shunt serves as an alternate route to convert alpha-ketoglutarate into succinate, bypassing the TCA cycle intermediate succinyl-CoA and instead producing the intermediate GABA. Transamination and subsequent decarboxylation of alpha-ketoglutarate leads to the formation of GABA. GABA is then metabolized by GABA transaminase to succinic semialdehyde. Finally, succinic semialdehyde is oxidized by succinic semialdehyde dehydrogenase (SSADH) to form succinate, re-entering the TCA cycle and closing the loop. Enzymes required for the GABA shunt are expressed in neurons, glial cells, macrophages and pancreatic cells.
Metabolic intermediate
Succinate is produced and concentrated in the mitochondria and its primary biological function is that of a metabolic intermediate. All metabolic pathways that are interlinked with the TCA cycle, including the metabolism of carbohydrates, amino acids, fatty acids, cholesterol, and heme, rely on the temporary formation of succinate. The intermediate is made available for biosynthetic processes through multiple pathways, including the reductive branch of the TCA cycle or the glyoxylate cycle, which are able to drive net production of succinate. In rodents, mitochondrial concentrations are approximately ~0.5 mM while plasma concentration are only 2–20 μM.
ROS production
The activity of succinate dehydrogenase (SDH), which interconverts succinate into fumarate participates in mitochondrial reactive oxygen species (ROS) production by directing electron flow in the electron transport chain. Under conditions of succinate accumulation, rapid oxidation of succinate by SDH can drive reverse electron transport (RET). If mitochondrial respiratory complex III is unable to accommodate excess electrons supplied by succinate oxidation, it forces electrons to flow backwards along the electron transport chain. RET at mitochondrial respiratory complex 1, the complex normally preceding SDH in the electron transport chain, leads to ROS production and creates a pro-oxidant microenvironment.
Additional biologic functions
In addition to its metabolic roles, succinate serves as an intracellular and extracellular signaling molecule. Extra-mitochondrial succinate alters the epigenetic landscape by inhibiting the family of 2-oxogluterate-dependent dioxygenases. Alternative, succinate can be released into the extracellular milieu and the blood stream where it is recognized by target receptors. In general, leakage from the mitochondria requires succinate overproduction or underconsumption and occurs due to reduced, reverse or completely absent activity of SDH or alternative changes in metabolic state. Mutations in SDH, hypoxia or energetic misbalance are all linked to an alteration of flux through the TCA cycle and succinate accumulation. Upon exiting the mitochondria, succinate serves as a signal of metabolic state, communicating to neighboring cells how metabolically active the originating cell population is. As such, succinate links TCA cycle dysfunction or metabolic changes to cell-cell communication and to oxidative stress-related responses.
Succinate transporters
Succinate requires specific transporters to move through both the mitochondrial and plasma membrane. Succinate exits the mitochondrial matrix and passes through the inner mitochondrial membrane via dicarboxylate transporters, primarily SLC25A10, a succinate-fumarate/malate transporter. In the second step of mitochondrial export, succinate readily crosses the outer mitochondrial membrane through porins, nonspecific protein channels that facilitate the diffusion of molecules less than 1.5 kDa. Transport across the plasma membrane is likely tissue specific. A key candidate transporter is INDY (I'm not dead yet), a sodium-independent anion exchanger, which moves both dicarboxylate and citrate into the bloodstream.
Extracellular signaling
Extracellular succinate can act as a signaling molecule with hormone-like function, targeting a variety of tissues such as blood cells, adipose tissue, immune cells, the liver, the heart, the retina and primarily the kidney. The G-protein coupled receptor, GPR91 also known as SUCNR1, serves as the detector of extracellular succinate. Arg99, His103, Arg252, and Arg281 near the center of the receptor generate a positively charged binding site for succinate. The ligand specificity of GPR91 was rigorously tested using 800 pharmacologically active compounds and 200 carboxylic acid and succinate-like compounds, all of which demonstrated significantly lower binding affinity. Overall, the EC50 for succinate-GPR91 is in the 20–50 uM range. Depending on the cell type, GPR91 can interact with multiple G proteins, including Gs, Gi and Gq, and enabling a multitude of signaling outcomes.
Effect on adipocytes
In adipocytes, the succinate-activated GPR91 signaling cascade inhibits lipolysis.
Effect on the liver and retina
Succinate signaling often occurs in response to hypoxic conditions. In the liver, succinate serves as a paracrine signal, released by anoxic hepatocytes, and targets stellate cells via GPR91. This leads to stellate cell activation and fibrogenesis. Thus, succinate is thought to play a role in liver homeostasis. In the retina, succinate accumulates in retinal ganglion cells in response to ischemic conditions. Autocrine succinate signaling promotes retinal neovascularization, triggering the activation of angiogenic factors such as endothelial growth factor (VEGF).
Effect on the heart
Extracellular succinate regulates cardiomyocyte viability through GPR91 activation; long-term succinate exposure leads to pathological cardiomyocyte hypertrophy. Stimulation of GPR91 triggers at least two signaling pathways in the heart: a MEK1/2 and ERK1/2 pathway that activates hypertrophic gene expression and a phospholipase C pathway which changes the pattern of Ca2+ uptake and distribution and triggers CaM-dependent hypertrophic gene activation.
Effect on immune cells
SUCNR1 is highly expressed on immature dendritic cells, where succinate binding stimulates chemotaxis. Furthermore, SUCNR1 synergizes with toll-like receptors to increase the production of proinflammatory cytokines such as TNF alpha and interleukin-1beta. Succinate may enhance adaptive immunity by triggering the activity of antigen-presenting cells that, in turn, activate T-cells.
Effect on platelets
SUCNR1 is one of the highest expressed G protein-coupled receptors on human platelets, present at levels similar to P2Y12, though the role of succinate signaling in platelet aggregation is debated. Multiple studies have demonstrated succinate-induced aggregation, but the effect has high inter-individual variability.
Effect on the kidneys
Succinate serves as a modulator of blood pressure by stimulating renin release in macula densa and juxtaglomerular apparatus cells via GPR91. Therapies targeting succinate to reduce cardiovascular risk and hypertension are currently under investigation.
Intracellular signaling
Accumulation of either fumarate or succinate reduces the activity of 2-oxogluterate-dependent dioxygenases, including histone and DNA demethylases, prolyl hydroxylases and collagen prolyl-4-hydroxyalses, through competitive inhibition. 2-oxoglutarate-dependent dioxygenases require an iron cofactor to catalyze hydroxylations, desaturations and ring closures. Simultaneous to substrate oxidation, they convert 2-oxglutarate, also known as alpha-ketoglutarate, into succinate and CO2. 2-oxoglutarate-dependent dioxygenases bind substrates in a sequential, ordered manner. First, 2-oxoglutarate coordinates with an Fe(II) ion bound to a conserved 2-histidinyl–1-aspartyl/glutamyl triad of residues present in enzymatic center. Subsequently, the primary substrate enters the binding pocket and lastly dioxygen binds to the enzyme-substrate complex. Oxidative decarboxylation then generates a ferryl intermediate coordinated to succinate, which serves to oxidize the bound primary substrate. Succinate may interfere with the enzymatic process by attaching to the Fe(II) center first, prohibiting the binding of 2-oxoglutarate. Thus, via enzymatic inhibition, increased succinate load can lead to changes in transcription factor activity and genome-wide alterations in histone and DNA methylation.
Epigenetic effects
Succinate and fumarate inhibit the TET (ten-eleven translocation) family of 5-methylcytosine DNA modifying enzymes and the JmjC domain-containing histone lysine demethylase (KDM). Pathologically elevated levels of succinate lead to hypermethylation, epigenetic silencing and changes in neuroendocrine differentiation, potentially driving cancer formation.
Gene regulation
Succinate inhibition of prolyl hydroxylases (PHDs) stabilizes the transcription factor hypoxia inducible factor (HIF)1α. PHDs hydroxylate proline in parallel to oxidatively decarboxylating 2-oxyglutarate to succinate and CO2. In humans, three HIF prolyl 4-hydroxylases regulate the stability of HIFs. Hydroxylation of two prolyl residues in HIF1α facilitates ubiquitin ligation, thus marking it for proteolytic destruction by the ubiquitin/proteasome pathway. Since PDHs have an absolute requirement for molecular oxygen, this process is suppressed in hypoxia allowing HIF1α to escape destruction. High concentrations of succinate will mimic the hypoxia state by suppressing PHDs, therefore stabilizing HIF1α and inducing the transcription of HIF1-dependent genes even under normal oxygen conditions. HIF1 is known to induce transcription of more than 60 genes, including genes involved in vascularization and angiogenesis, energy metabolism, cell survival, and tumor invasion.
Inflammation
Metabolic signaling involving succinate can be involved in inflammation via stabilization of HIF1-alpha or GPR91 signaling in innate immune cells. Through these mechanisms, succinate accumulation has been shown to regulate production of inflammatory cytokines. For dendritic cells, succinate functions as a chemoattractant and increases their antigen-presenting function via receptor stimulated cytokine production. In inflammatory macrophages, succinate-induced stability of HIF1 results in increased transcription of HIF1-dependent genes in spite of hypoxic conditions; including the pro-inflammatory cytokine interleukin-1β. Other inflammatory cytokines produced by activated macrophages such as tumor necrosis factor or interleukin 6 are not directly affected by succinate and HIF1. The mechanism by which succinate accumulates in immune cells is not fully understood. Activation of inflammatory macrophages through toll-like receptors induces a metabolic shift towards glycolysis. In spite of a general downregulation of the TCA cycle under these conditions, succinate concentration is increased. However, lipopolysaccharides involved in the activation of macrophages increase glutamine and GABA transporters. Succinate may thus be produced from enhanced glutamine metabolism via alpha-ketoglutarate or the GABA shunt.
Tumorigenesis
Succinate is one of three oncometabolites, metabolic intermediates whose accumulation causes metabolic and non-metabolic dysregulation implicated in tumorigenesis. Loss-of-function mutations in the genes encoding succinate dehydrogenase, frequently found in hereditary paraganglioma and pheochromocytoma, cause pathological increase in succinate. SDH mutations have also been identified in gastrointestinal stromal tumors, renal tumors, thyroid tumors, testicular seminomas and neuroblastomas. The oncogenic mechanism caused by mutated SHD is thought to relate to succinate's ability to inhibit 2-oxogluterate-dependent dioxygenases. Inhibition of KDMs and TET hydroxylases results in epigenetic dysregulation and hypermethylation affecting genes involved in cell differentiation. Additionally, succinate-promoted activation of HIF-1α generates a pseudo-hypoxic state that can promote tumorneogensis by transcriptional activation of genes involved in proliferation, metabolism and angiogenesis. The other two oncometabolites, fumarate and 2-hydroxyglutarate have similar structures to succinate and function through parallel HIF-inducing oncogenic mechanisms.
Ischemia reperfusion injury
Succinate accumulation under hypoxic conditions has been implicated in the reperfusion injury through increased ROS production. During ischemia, fumarate is formed from purine nucleotide breakdown and partial reversal of the malate/aspartate shuttle. Fumarate overflow results in the production and accumulation of succinate through reverse activity of SDH. Upon reperfusion, succinate is rapidly oxidized leading to abrupt and extensive production of ROS. ROS then trigger the cellular apoptotic machinery or induce oxidative damage to proteins, membranes, organelles etc. In animal models, pharmacological inhibition of ischemic succinate accumulation ameliorated ischemia-reperfusion injury. The inhibition of succinate-mediated ROS production is currently under investigation as a therapeutic target.