
Specialty oncology DiseasesDB 33480 | ICD-O M8680/0 - M8693/9 eMedicine med/2994 | |
 | ||
ICD-10 C75.4, C75.5, D35.5, D35.6, D44.6, D44.7 ICD-9-CM 194.5, 194.6, 227.5, 227.6, 237.3 | ||
A paraganglioma is a rare neuroendocrine neoplasm that may develop at various body sites (including the head, neck, thorax and abdomen). Unlike other types of cancer, there is no test that determines benign from malignant tumors, long term followup is therefore recommended for all individuals with paraganglioma. Approximately 50% of patients with recurrent disease experience distant metastasis. The 5-year survival in the setting of metastatic disease is 40% to 45%. (https://www.cancer.gov/types/pheochromocytoma/patient/pheochromocytoma-treatment-pdq)
Contents
Cellular origin and classification
Paragangliomas originate from paraganglia in chromaffin-negative glomus cells derived from the embryonic neural crest, functioning as part of the sympathetic nervous system (a branch of the autonomic nervous system). These cells normally act as special chemoreceptors located along blood vessels, particularly in the carotid bodies (at the bifurcation of the common carotid artery in the neck) and in aortic bodies (near the aortic arch).
Accordingly, paragangliomas are categorised as originating from a neural cell line in the World Health Organization classification of neuroendocrine tumors. In the categorization proposed by Wick, paragangliomas belong to group II. Given the fact that they originate from cells of the orthosympathetic system, paragangliomas are closely related to pheochromocytomas, which however are chromaffin-positive.
Clinical presentation
Most paragangliomas are either asymptomatic or present as a painless mass. While all contain neurosecretory granules, only in 1–3% of cases is secretion of hormones such as catecholamines abundant enough to be clinically significant; in that case manifestations often resemble those of pheochromocytomas (intra-medullary paraganglioma).
Inheritance
About 75% of paragangliomas are sporadic; the remaining 25% are hereditary (and have an increased likelihood of being multiple and of developing at an earlier age). Mutations of the genes for the succinate dehydrogenase, SDHD (previously known as PGL1), SDHA, SDHC (previously PGL3) and SDHB have been identified as causing familial head and neck paragangliomas. Mutations of SDHB play an important role in familial adrenal pheochromocytoma and extra-adrenal paraganglioma (of abdomen and thorax), although there is considerable overlap in the types of tumors associated with SDHB and SDHD gene mutations. Paragangliomas may also occur in MEN type 2A and 2B. They are seen in at a higher incidence in people living at high altitude. Other genes related to familiar paraganglioma are SDHAF2, VHL, NF1, TMEM127 and MAX.
Pathology
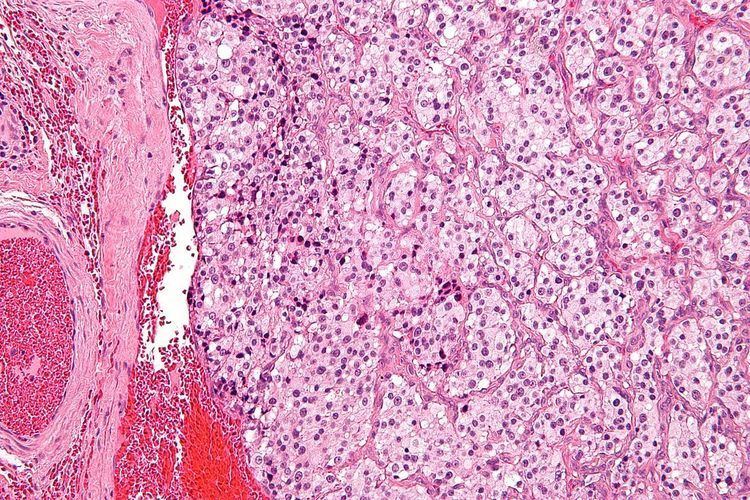
The paragangliomas appear grossly as sharply circumscribed polypoid masses and they have a firm to rubbery consistency. They are highly vascular tumors and may have a deep red color.
On microscopic inspection, the tumor cells are readily recognized. Individual tumor cells are polygonal to oval and are arranged in distinctive cell balls, called Zellballen. These cell balls are separated by fibrovascular stroma and surrounded by sustentacular cells.
By light microscopy, the differential diagnosis includes related neuroendocrine tumors, such as carcinoid tumor, neuroendocrine carcinoma, and medullary carcinoma of the thyroid.
With immunohistochemistry, the chief cells located in the cell balls are positive for chromogranin, synaptophysin, neuron specific enolase, serotonin, neurofilament and Neural cell adhesion molecule; they are S-100 protein negative. The sustentacular cells are S-100 positive and focally positive for glial fibrillary acidic protein. By histochemistry, the paraganglioma cells are argyrophilic, periodic acid Schiff negative, mucicarmine negative, and argentaffin negative.
Sites of origin
About 85% of paragangliomas develop in the abdomen; only 12% develop in the chest and 3% in the head and neck region (the latter are the most likely to be symptomatic). While most are single, rare multiple cases occur (usually in a hereditary syndrome). Paragangliomas are described by their site of origin and are often given special names:-
Treatment
The main treatment modalities are surgery, embolization and radiotherapy.