
Pronunciation /ˈɛnsɛd/ EN-sed ATC code M01A Biological target COX-1 and COX-2 | ||
 | ||
Synonyms nonsteroidal anti-inflammatory agents/analgesics (NSAIAs), nonsteroidal anti-inflammatory medicines (NSAIMs) | ||
Nsaid pharmacology an introduction
Nonsteroidal anti-inflammatory drugs (NSAIDs) are a drug class that groups together drugs that provide analgesic (Pain-killing) and antipyretic (fever-reducing) effects, and, in higher doses, anti-inflammatory effects.
Contents
- Nsaid pharmacology an introduction
- 11 pm1 moa of nsaids
- Medical uses
- Contraindications
- Adverse effects
- Combinational risk
- Cardiovascular
- Possible erectile dysfunction risk
- Gastrointestinal
- Inflammatory bowel disease
- Renal
- Photosensitivity
- During pregnancy
- Allergyallergy like hypersensitivity reactions
- Other
- Drug interactions
- Mechanism of action
- Antipyretic activity
- Classification
- Anthranilic acid derivatives Fenamates
- Sulfonanilides
- Others
- Chirality
- Main practical differences
- Pharmacokinetics
- History
- Research
- Veterinary use
- References
The term nonsteroidal distinguishes these drugs from steroids, which, among a broad range of other effects, have a similar eicosanoid-depressing, anti-inflammatory action. First used in 1960, the term served to distance new drugs from steroid-related iatrogenic tragedies.
The most prominent members of this group of drugs are aspirin, ibuprofen and naproxen, all available over the counter in most countries. Paracetamol (acetaminophen) is generally not considered an NSAID because it has only little anti-inflammatory activity. It treats pain mainly by blocking COX-2 mostly in the central nervous system, but not much in the rest of the body.
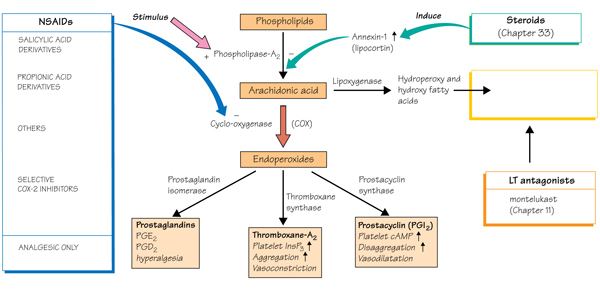
Most NSAIDs inhibit the activity of cyclooxygenase-1 (COX-1) and cyclooxygenase-2 (COX-2), and thereby the synthesis of prostaglandins and thromboxanes. It is thought that inhibiting COX-2 leads to the anti-inflammatory, analgesic and antipyretic effects and that those NSAIDs also inhibiting COX-1, particularly Aspirin, may cause gastrointestinal bleeding and ulcers.
11 pm1 moa of nsaids
Medical uses
NSAIDs are usually used for the treatment of acute or chronic conditions where pain and inflammation are present.
NSAIDs are generally used for the symptomatic relief of the following conditions:

Aspirin, the only NSAID able to irreversibly inhibit COX-1, is also indicated for inhibition of platelet aggregation. This is useful for the management of arterial thrombosis and prevention of adverse cardiovascular events. Aspirin inhibits platelet aggregation by inhibiting the action of thromboxane A2.
NSAIDs are useful in the management of post-operative dental pain following invasive dental procedures such as dental extraction. When not contra-indicated they are favoured over the use of paracetamol alone due to the anti-inflammatory effect they provide. When used in combination with paracetamol the analgesic effect has been proven to be improved. A 2012 Cochrane review found that there is some weak evidence suggesting that taking pre-operative analgesia can reduce the length of post operative pain associated with placing orthodontic spacers under local anaesthetic.
Contraindications
NSAIDs may be used with caution by people with the following conditions:

NSAIDs should usually be avoided by people with the following conditions:
Adverse effects
The widespread use of NSAIDs has meant that the adverse effects of these drugs have become increasingly common. Use of NSAIDs increases risk of having a range of gastrointestinal (GI) problems. When NSAIDs are used for pain management after surgery they cause increased risk of kidney problems.
An estimated 10–20% of NSAID patients experience dyspepsia. In the 1990s high doses of prescription NSAIDs were associated with serious upper gastrointestinal adverse events, including bleeding. Over the past decade, deaths associated with gastric bleeding have declined.
NSAIDs, like all drugs, may interact with other medications. For example, concurrent use of NSAIDs and quinolones may increase the risk of quinolones' adverse central nervous system effects, including seizure.
There is an argument over the benefits and risks of NSAIDs for treating chronic musculoskeletal pain. Each drug has a benefit-risk profile and balancing the risk of no treatment with the competing potential risks of various therapies is the clinician's responsibility.
Combinational risk
If a COX-2 inhibitor is taken, a traditional NSAID (prescription or over-the-counter) should not be taken at the same time. In addition, people on daily aspirin therapy (e.g., for reducing cardiovascular risk) must be careful if they also use other NSAIDs, as these may inhibit the cardioprotective effects of aspirin.
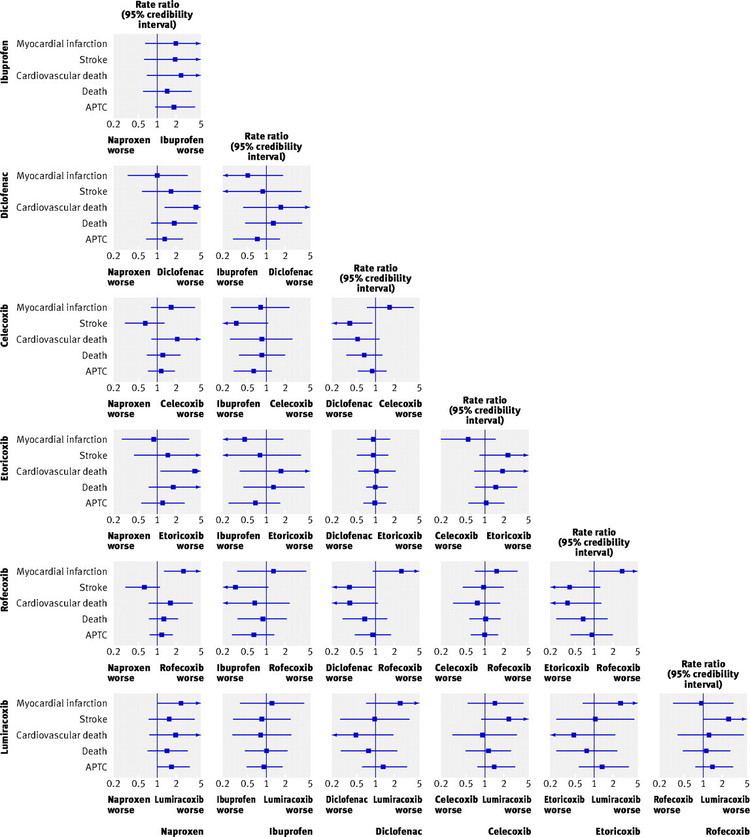
Rofecoxib (Vioxx) was shown to produce significantly fewer gastrointestinal adverse drug reactions (ADRs) compared with naproxen. This study, the VIGOR trial, raised the issue of the cardiovascular safety of the coxibs. A statistically significant increase in the incidence of myocardial infarctions was observed in patients on Rofecoxib. Further data, from the APPROVe trial, showed a statistically significant relative risk of cardiovascular events of 1.97 versus placebo—which caused a worldwide withdrawal of rofecoxib in October 2004.
Use of methotrexate together with NSAIDS in Rheumatoid arthritis is safe, if adequate monitoring is done.
Cardiovascular
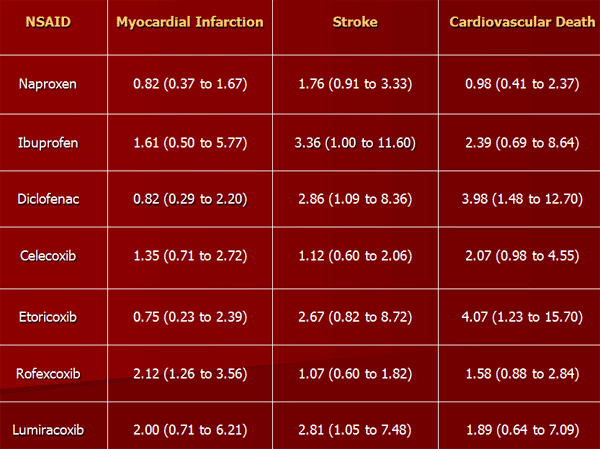
NSAIDs aside from aspirin, both newer selective COX-2 inhibitors and traditional anti-inflammatories, increase the risk of myocardial infarction and stroke. They are not recommended in those who have had a previous heart attack as they increase the risk of death and/or recurrent MI. Evidence indicates that naproxen may be the least harmful out of these.
NSAIDs aside from (low-dose) aspirin are associated with a doubled risk of heart failure in people without a history of cardiac disease. In people with such a history, use of NSAIDs (aside from low-dose aspirin) was associated with a more than 10-fold increase in heart failure. If this link is proven causal, researchers estimate that NSAIDs would be responsible for up to 20 percent of hospital admissions for congestive heart failure. In people with heart failure, NSAIDs increase mortality risk (hazard ratio) by approximately 1.2–1.3 for naproxen and ibuprofen, 1.7 for rofecoxib and Celecoxib, and 2.1 for diclofenac.
On 9 July 2015, the FDA toughened warnings of increased heart attack and stroke risk associated with nonsteroidal anti-inflammatory drugs (NSAID). Aspirin is an NSAID but is not affected by the new warnings.
Possible erectile dysfunction risk
A 2005 Finnish study linked long term (over 3 months) use of NSAIDs with an increased risk of erectile dysfunction. This study was correlational only, and depended solely on self-reports (questionnaires).
A 2011 publication in The Journal of Urology received widespread publicity. According to this study, men who used NSAIDs regularly were at significantly increased risk of erectile dysfunction. A link between NSAID use and erectile dysfunction still existed after controlling for several conditions. However, the study was observational and not controlled, with low original participation rate, potential participation bias, and other uncontrolled factors. The authors warned against drawing any conclusion regarding cause.
Gastrointestinal
The main adverse drug reactions (ADRs) associated with NSAID use relate to direct and indirect irritation of the gastrointestinal (GI) tract. NSAIDs cause a dual assault on the GI tract: the acidic molecules directly irritate the gastric mucosa, and inhibition of COX-1 and COX-2 reduces the levels of protective prostaglandins. Inhibition of prostaglandin synthesis in the GI tract causes increased gastric acid secretion, diminished bicarbonate secretion, diminished mucus secretion and diminished trophic effects on the epithelial mucosa.
Common gastrointestinal ADRs include:
Clinical NSAID ulcers are related to the systemic effects of NSAID administration. Such damage occurs irrespective of the route of administration of the NSAID (e.g., oral, rectal, or parenteral) and can occur even in patients with achlorhydria.
Ulceration risk increases with therapy duration, and with higher doses. To minimize GI ADRs, it is prudent to use the lowest effective dose for the shortest period of time—a practice that studies show is often not followed. Recent studies show that over 50% of patients who take NSAIDs have sustained some mucosal damage to their small intestine.
There are also some differences in the propensity of individual agents to cause gastrointestinal ADRs. Indomethacin, ketoprofen and piroxicam appear to have the highest prevalence of gastric ADRs, while ibuprofen (lower doses) and diclofenac appear to have lower rates.
Certain NSAIDs, such as aspirin, have been marketed in enteric-coated formulations that manufacturers claim reduce the incidence of gastrointestinal ADRs. Similarly, some believe that rectal formulations may reduce gastrointestinal ADRs. However, consistent with the systemic mechanism of such ADRs, and in clinical practice, these formulations have not demonstrated a reduced risk of GI ulceration.
Commonly, gastric (but not necessarily intestinal) adverse effects can be reduced through suppressing acid production, by concomitant use of a proton pump inhibitor, e.g., omeprazole, esomeprazole, or the prostaglandin analog misoprostol. Misoprostol is itself associated with a high incidence of gastrointestinal ADRs (diarrhea). While these techniques may be effective, they are expensive for maintenance therapy.
Inflammatory bowel disease
NSAIDs should be used with caution in individuals with inflammatory bowel disease (e.g., Crohn's disease or ulcerative colitis) due to their tendency to cause gastric bleeding and form ulceration in the gastric lining. Pain relievers such as paracetamol (also known as acetaminophen) or drugs containing codeine (which slows down bowel activity) are safer medications for pain relief in IBD.
Renal
NSAIDs are also associated with a fairly high incidence of renal adverse drug reactions (ADRs). The mechanism of these renal ADRs is due to changes in renal hemodynamics (kidney blood flow), ordinarily mediated by prostaglandins, which are affected by NSAIDs. Prostaglandins normally cause vasodilation of the afferent arterioles of the glomeruli. This helps maintain normal glomerular perfusion and glomerular filtration rate (GFR), an indicator of renal function. This is particularly important in renal failure where the kidney is trying to maintain renal perfusion pressure by elevated angiotensin II levels. At these elevated levels, angiotensin II also constricts the afferent arteriole into the glomerulus in addition to the efferent arteriole it normally constricts. Prostaglandins serve to dilate the afferent arteriole; by blocking this prostaglandin-mediated effect, particularly in renal failure, NSAIDs cause unopposed constriction of the afferent arteriole and decreased RPF (renal perfusion pressure).
Common ADRs associated with altered renal function include:
These agents may also cause kidney impairment, especially in combination with other nephrotoxic agents. Kidney failure is especially a risk if the patient is also concomitantly taking an ACE inhibitor (which removes angiotensin II's vasoconstriction of the efferent arteriole) and a diuretic (which drops plasma volume, and thereby RPF)—the so-called "triple whammy" effect.
In rarer instances NSAIDs may also cause more severe kidney conditions:
NSAIDs in combination with excessive use of phenacetin and/or paracetamol (acetaminophen) may lead to analgesic nephropathy.
Photosensitivity
Photosensitivity is a commonly overlooked adverse effect of many of the NSAIDs. The 2-arylpropionic acids are the most likely to produce photosensitivity reactions, but other NSAIDs have also been implicated including piroxicam, diclofenac and benzydamine.
Benoxaprofen, since withdrawn due to its hepatotoxicity, was the most photoactive NSAID observed. The mechanism of photosensitivity, responsible for the high photoactivity of the 2-arylpropionic acids, is the ready decarboxylation of the carboxylic acid moiety. The specific absorbance characteristics of the different chromophoric 2-aryl substituents, affects the decarboxylation mechanism. While ibuprofen has weak absorption, it has been reported as a weak photosensitising agent.
During pregnancy
NSAIDs are not recommended during pregnancy, particularly during the third trimester. While NSAIDs as a class are not direct teratogens, they may cause premature closure of the fetal ductus arteriosus and renal ADRs in the fetus. Additionally, they are linked with premature birth and miscarriage. Aspirin, however, is used together with heparin in pregnant women with antiphospholipid antibodies. Additionally, Indomethacin is used in pregnancy to treat polyhydramnios by reducing fetal urine production via inhibiting fetal renal blood flow.
In contrast, paracetamol (acetaminophen) is regarded as being safe and well-tolerated during pregnancy, but Leffers et al. released a study in 2010 indicating that there may be associated male infertility in the unborn. Doses should be taken as prescribed, due to risk of hepatotoxicity with overdoses.
In France, the country's health agency contraindicates the use of NSAIDs, including aspirin, after the sixth month of pregnancy.
Allergy/allergy-like hypersensitivity reactions
A variety of allergic or allergic-like NSAID hypersensitivity reactions follow the ingestion of NSAIDs. These hypersensitivity reactions differ from the other adverse reactions listed here which are toxicity reactions, i.e. unwanted reactions that result from the pharmacological action of a drug, are dose-related, and can occur in any treated individual; hypersensitivity reactions are idiosyncratic reactions to a drug. Some NSAID hypersensitivity reactions are truly allergic in origin: 1) repetitive IgE-mediated urticarial skin eruptions, angioedema, and anaphylaxis following immediately to hours after ingesting one structural type of NSAID but not after ingesting structurally unrelated NSAIDs; and 2) Comparatively mild to moderately severe T cell-mediated delayed onset (usually more than 24 hour), skin reactions such as maculopapular rash, fixed drug eruptions, photosensitivity reactions, delayed urticaria, and contact dermatitis; or 3) far more severe and potentially life-threatening t-cell-mediated delayed systemic reactions such as the DRESS syndrome, acute generalized exanthematous pustulosis, the Stevens–Johnson syndrome, and toxic epidermal necrolysis. Other NSAID hypersensitivity reactions are allergy-like symptoms but do not involve true allergic mechanisms; rather, they appear due to the ability of NSAIDs to alter the metabolism of arachidonic acid in favor of forming metabolites that promote allergic symptoms. Afflicted individuals may be abnormally sensitive to these provocative metabolites and/or overproduce them and typically are susceptible to a wide range of structurally dissimilar NSAIDs, particularly those that inhibit COX1. Symptoms, which develop immediately to hours after ingesting any of various NSAIDs that inhibit COX-1, are: 1) exacerbations of asthmatic and rhinitis (see aspirin-induced asthma) symptoms in individuals with a history of asthma or rhinitis and 2) exacerbation or first-time development of wheals and/or angioedema in individuals with or without a history of chronic urticarial lesions or angioedema.
Other
Common adverse drug reactions (ADR), other than listed above, include: raised liver enzymes, Headache, dizziness. Uncommon ADRs include: hyperkalaemia, confusion, bronchospasm, rash. Rapid and severe swelling of the face and/or body. Ibuprofen may also rarely cause irritable bowel syndrome symptoms. NSAIDs are also implicated in some cases of Stevens–Johnson syndrome.
Most NSAIDs penetrate poorly into the central nervous system (CNS). However, the COX enzymes are expressed constitutively in some areas of the CNS, meaning that even limited penetration may cause adverse effects such as somnolence and dizziness.
In very rare cases, ibuprofen can cause aseptic meningitis.
As with other drugs, allergies to NSAIDs might exist. While many allergies are specific to one NSAID, up to 1 in 5 people may have unpredictable cross-reactive allergic responses to other NSAIDs as well.
Drug interactions
NSAIDs reduce renal blood flow and thereby decrease the efficacy of diuretics, and inhibit the elimination of lithium and methotrexate.
NSAIDs cause hypocoagulability, which may be serious when combined with other drugs that also decrease blood clotting, such as warfarin.
NSAIDs may aggravate Hypertension (high blood pressure) and thereby antagonize the effect of antihypertensives, such as ACE inhibitors.
NSAIDs may interfere and reduce efficiency of SSRI antidepressants.
Various widely used nonsteroidal anti-inflammatory drugs (NSAIDs) enhance endocannabinoid signaling by blocking the anandamide-degrading membrane enzyme fatty acid amide hydrolase (FAAH).
Mechanism of action
Most NSAIDs act as nonselective inhibitors of the enzyme cyclooxygenase (COX), inhibiting both the cyclooxygenase-1 (COX-1) and cyclooxygenase-2 (COX-2) isoenzymes. This inhibition is competitively reversible (albeit at varying degrees of reversibility), as opposed to the mechanism of aspirin, which is irreversible inhibition. COX catalyzes the formation of prostaglandins and thromboxane from Arachidonic acid (itself derived from the cellular phospholipid bilayer by phospholipase A2). Prostaglandins act (among other things) as messenger molecules in the process of inflammation. This mechanism of action was elucidated by John Vane (1927–2004), who received a Nobel Prize for his work (see Mechanism of action of aspirin).
COX-1 is a constitutively expressed enzyme with a "house-keeping" role in regulating many normal physiological processes. One of these is in the stomach lining, where prostaglandins serve a protective role, preventing the stomach mucosa from being eroded by its own acid. COX-2 is an enzyme facultatively expressed in inflammation, and it is inhibition of COX-2 that produces the desirable effects of NSAIDs.
When nonselective COX-1/COX-2 inhibitors (such as aspirin, ibuprofen, and naproxen) lower stomach prostaglandin levels, ulcers of the stomach or duodenum internal bleeding can result.
NSAIDs have been studied in various assays to understand how they affect each of these enzymes. While the assays reveal differences, unfortunately, different assays provide differing ratios.
The discovery of COX-2 led to research to the development of selective COX-2 inhibiting drugs that do not cause gastric problems characteristic of older NSAIDs.
Paracetamol (acetaminophen) is not considered an NSAID because it has little anti-inflammatory activity. It treats pain mainly by blocking COX-2 mostly in the central nervous system, but not much in the rest of the body.
However, many aspects of the Mechanism of action of NSAIDs remain unexplained, and for this reason, further COX pathways are hypothesized. The COX-3 pathway was believed to fill some of this gap but recent findings make it appear unlikely that it plays any significant role in humans and alternative explanation models are proposed.
NSAIDs interact with the endocannabinoid system and its endocannabinoids, as COX2 have been shown to utilize endocannabinoids as substrates, and may have a key role in both the therapeutic and adverse effects of NSAIDs, as well as in NSAIDs-induced placebo responses.
NSAIDs are also used in the acute pain caused by gout because they inhibit urate crystal phagocytosis besides inhibition of prostaglandin synthase.
Antipyretic activity
NSAIDS have antipyretic activity and can be used to treat fever. Fever is caused by elevated levels of prostaglandin E2, which alters the firing rate of neurons within the hypothalamus that control thermoregulation. Antipyretics work by inhibiting the enzyme COX, which causes the general inhibition of prostanoid biosynthesis (PGE2) within the hypothalamus. PGE2 signals to the hypothalamus to increase the body's thermal set point. Ibuprofen has been shown more effective as an antipyretic than paracetamol (acetaminophen). Arachidonic acid is the precursor substrate for cyclooxygenase leading to the production of prostaglandins F, D & E.
Classification
NSAIDs can be classified based on their chemical structure or mechanism of action. Older NSAIDs were known long before their mechanism of action was elucidated and were for this reason classified by chemical structure or origin. Newer substances are more often classified by mechanism of action.
Anthranilic acid derivatives (Fenamates)
The following NSAIDs are derived from fenamic acid. which is a derivative of anthranilic acid, which in turn is a nitrogen isostere of Salicylic acid, which is the active metabolite of aspirin.
Sulfonanilides
Others
Chirality
Most NSAIDs are chiral molecules (diclofenac is a notable exception). However, the majority are prepared in a racemic mixture. Typically, only a single enantiomer is pharmacologically active. For some drugs (typically profens), an isomerase enzyme in vivo converts the inactive enantiomer into the active form, although its activity varies widely in individuals. This phenomenon is likely responsible for the poor correlation between NSAID efficacy and plasma concentration observed in older studies, when specific analysis of the active enantiomer was not performed.
Ibuprofen and Ketoprofen are now available in single, active enantiomer preparations (Dexibuprofen and Dexketoprofen), which purport to offer quicker onset and an improved side-effect profile. Naproxen has always been marketed as the single active enantiomer.
Main practical differences
NSAIDs within a group tend to have similar characteristics and tolerability. There is little difference in clinical efficacy among the NSAIDs when used at equivalent doses. Rather, differences among compounds usually relate to dosing regimens (related to the compound's elimination half-life), route of administration, and tolerability profile.
Regarding adverse effects, selective COX-2 inhibitors have lower risk of gastrointestinal bleeding, and there is no significant increase in risk of myocardial infarction. With the exception of naproxen, nonselective NSAIDs increase the risk of having a heart attack. Some data also supports that the partially selective Nabumetone is less likely to cause gastrointestinal events.
A consumer report noted that ibuprofen, naproxen, and Salsalate are less expensive than other NSAIDs, and essentially as effective and safe when used appropriately to treat Osteoarthritis and pain.
Pharmacokinetics
Most nonsteroidal anti-inflammatory drugs are weak acids, with a pKa of 3–5. They are absorbed well from the stomach and intestinal mucosa. They are highly protein-bound in plasma (typically >95%), usually to albumin, so that their volume of distribution typically approximates to plasma volume. Most NSAIDs are metabolized in the liver by oxidation and conjugation to inactive metabolites that typically are excreted in the urine, though some drugs are partially excreted in bile. Metabolism may be abnormal in certain disease states, and accumulation may occur even with normal dosage.
Ibuprofen and diclofenac have short half-lives (2–3 hours). Some NSAIDs (typically oxicams) have very long half-lives (e.g. 20–60 hours).
History
From the era of Greek medicine to the mid-19th century, the discovery of medicinal agents was classed as an empirical art; folklore and mythological guidance were combined in deploying the vegetable and mineral products that made up the expansive pharmacopeia of the time. Myrtle leaves were in use by 1500 BCE. Hippocrates (460–377 BCE) first reported using willow bark and in 30 BCE Celsus described the signs of inflammation and also used willow bark to mitigate them. On 25 April 1763, Edward Stone wrote to the Royal Society describing his observations on the use of willow bark-based medicines in febrile patients. The active ingredient of willow bark, a glycoside called salicin, was first isolated by Johann Andreas Buchner in 1827. By 1829, French chemist Henri Leroux had improved the extraction process to obtain about 30g of purified salicin from 1.5 kg of bark.
By hydrolysis, salicin releases glucose and salicylic alcohol which can be converted into salicylic acid, both in vivo and through chemical methods. The acid is more effective than salicin and, in addition to its fever-reducing properties, is anti-inflammatory and analgesic. In 1869, Hermann Kolbe synthesised salicylate, although it was too acidic for the gastric mucosa. The reaction used to synthesise aromatic acid from a phenol in the presence of CO2 is known as the Kolbe-Schmitt reaction.
By 1897 the German chemist Felix Hoffmann and the Bayer company prompted a new age of pharmacology by converting salicylic acid into acetylsalicylic acid—named aspirin by Heinrich Dreser. Other NSAIDs were developed from the 1950s forward. In 2001, NSAIDs accounted for 70,000,000 prescriptions and 30 billion over-the-counter doses sold annually in the United States.
Research
While studies have been conducted to see if various NSAIDs can improve behavior in transgenic mouse models of Alzheimer's disease and observational studies in humans have shown promise, there is no good evidence from randomized clinical trials that NSAIDs can treat or prevent Alzheimer's in humans; clinical trials of NSAIDs for treatment of Alzheimer's have found more harm than benefit. Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) coordinate with Metal ions affecting the cellular function
Veterinary use
Research supports the use of NSAIDs for the control of pain associated with veterinary procedures such as dehorning and castration of calves. The best effect is obtained by combining a short-term local anesthetic such as lidocaine with an NSAID acting as a longer term analgesic. However, as different species have varying reactions to different medications in the NSAID family, little of the existing research data can be extrapolated to animal species other than those specifically studied, and the relevant government agency in one area sometimes prohibits uses approved in other jurisdictions.
For example, ketoprofen's effects have been studied in horses more than in ruminants but, due to controversy over its use in racehorses, veterinarians who treat livestock in the United States more commonly prescribe flunixin meglumine, which, while labeled for use in such animals, is not indicated for post-operative pain.
In the United States, Meloxicam is approved for use only in canines, whereas (due to concerns about liver damage) it carries warnings against its use in cats except for one-time use during surgery. In spite of these warnings, meloxicam is frequently prescribed "off-label" for non-canine animals including cats and livestock species. In other countries, for example The European Union (EU), there is a label claim for use in cats.