
 | ||
In pharmacology, GABAA receptor positive allosteric modulators are positive allosteric modulator (PAM) molecules that increase the activity of the GABAA receptor protein in the vertebrate central nervous system. Unlike GABAA receptor agonists, GABAA PAMs do not bind at the same active site as the γ-Aminobutyric acid (GABA) neurotransmitter molecule: they affect the receptor by binding at a different site on the protein. This is called allosteric modulation.
Contents
- History
- Barbiturates
- Benzodiazepines
- Receptor
- Available agents
- Insomnia
- Anticonvulsant
- Sedation
- Benzodiazepine
- Analgesic
- Schizophrenia
- Depression
- Stroke
- Neurosteroids
- Barbituric acid
- Biosynthesis of neurosteroids
- References
GABA is a major inhibitory neurotransmitter in the central nervous system. Upon binding, it triggers the GABAA receptor to open its chloride channel to allow chlorine ions into the neuron, making the cell hyperpolarized and less likely to fire. GABAA PAMs bind increase the effect of GABA by making the channel open more frequently or for longer periods. However, they have no effect if GABA or another agonist is not present.
In psychopharmacology, GABAA receptor PAMs used as drugs have mainly sedative and anxiolytic effects. Examples of GABAA PAMs include alcohol (ethanol), benzodiazepines such as diazepam (Valium) and alprazolam (Xanax), Z-drugs such as zolpidem (Ambien) and the barbiturate drugs.
History
The GABAA receptors have historically been a target of drug treatment research. The earliest compounds were ions, such as bromide.
Barbiturates
In 1903, the first psychoactive derivative of barbituric acid was synthesized and marketed for headaches. Within 30 years, many other barbiturates were developed and found use as sedatives, sleep aids and general anesthetics. Although barbiturates fell out of favor, they continue to serve as a short-acting anesthetic and anti-epileptic drugs.
Benzodiazepines
Benzodiazepines were discovered in 1950 and largely replaced the barbiturates because of their larger therapeutic index. At first benzodiazepines were considered to be safe and efficient minor tranquilizers but then were criticized for their dependence producing effects. Several efficient benzodiazepines offer choices about dosage form, length of action, metabolic interaction and safety.
Benzodiazepines function by binding to the benzodiazepine site on most, but not all, GABAA receptors. GABAA modulation by benzodiazepine site agonists is self-limiting. The channel conductance is not higher in the presence of benzodiazepine and GABA than the conductance with the presence of only high GABA concentrations. Additionally, in the absence of GABA the presence of benzodiazepines alone does not open the chloride channel.
Certain metabolites of progesterone and deoxycorticosterone are potent and selective positive allosteric modulators of the γ-aminobutyric acid type A (GABAA) receptor. Hans Selye demonstrated in the 1940 that certain pregnane steroids could cause both anesthesia and sedation but 40 years later the molecular mechanism emerged to explain their depressant effect. In a rat brain slice preparation, the synthetic steroidal anesthetic alphaxalone (5α-pregnan-3α-ol-11,20 dione) enhanced both stimulus-evoked inhibition and the effects of exogenously applied muscimol which is a GABAA selective agonist.
Receptor
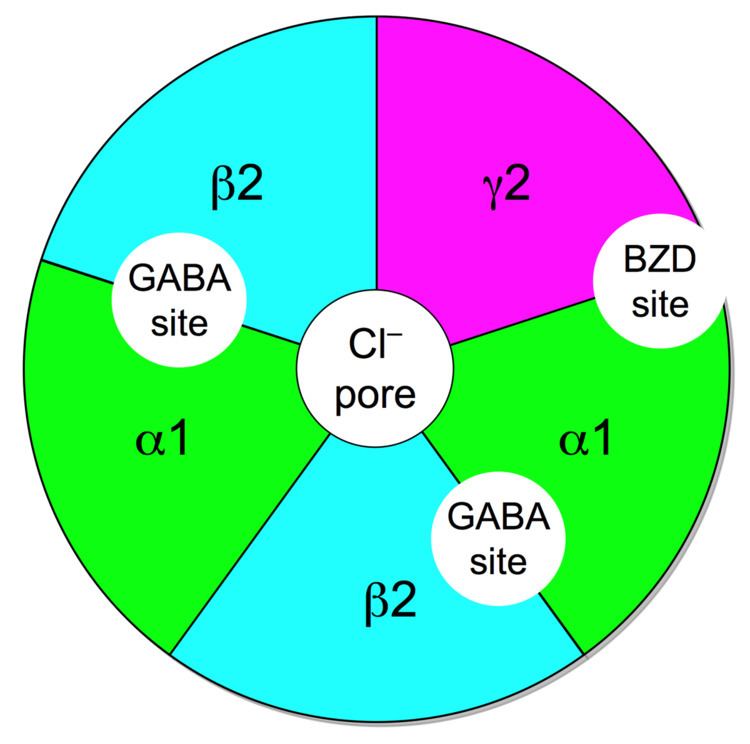
The GABAA receptors are made up of subunits which form a receptor complex. Humans have 19 receptor subunits and are classified into α (1–6), β (1–3), γ (1–3), δ, ε, π, θ, and ρ (1−3). The function of the receptor is different according to how the pentameric complex is put together. The most common complex that includes around 40% of the GABAA receptors is the α1β2γ2 combination. The expression of the subunits can be very different depending on brain region. The combination of the subunits influences how the receptor acts. For example, if the α1 and β2 subunits are expressed together they have high sensitivity to GABA, but low channel conductance. But if the γ2 is expressed with α1 and β2 the sensitivity is low and channel conductance is high. γ2 subunit has to be present for high affinity binding of benzodiazepine. Little is know about where different complexes are located in the brain, complicating drug discovery. For example, the binding site of neurosteroids in the GABAA receptor is not known and barbiturates bind at a beta subunit that is distinct from the benzodiazepine binding site.
Available agents
Barbiturates
Barbiturates' precise action sites have not yet been defined. The second and third transmembrane domains of the β subunit appear to be critical; binding may involve a pocket formed by β-subunit methionine 286 as well as α-subunit methionine 236.
Insomnia
Barbiturates were introduced as hypnotics for patients with schizophrenia. It induced a state of deep and prolonged sleep. But this was not used for long because of adverse side effects.
Anticonvulsant
Phenobarbital was the first truly effective drug against epilepsy. It was discovered by accident when given to epileptic patients to help them sleep. The positive side effects were anticonvulsant properties that reduced seizure number and intensity.
Sedation
Pentobarbital is used as a hypnotic when analgesia is not required. It´s often used in CT imaging when sedation is needed. It is efficient, safe and the recovery time is short. In 2013 the barbiturates phenobarbital and butabarbital are still used as sedatives in certain cases as well as to antagonize the effects of drugs as ephedrine and theophylline. Phenobarbital is used in cases of drug withdrawal syndromes. It is used as normal and emergency treatment in some cases of epilepsy.
Benzodiazepine
Synaptic action of benzodiazepines: GABAA receptors located at synapses are activated when they are exposed to high concentration of GABA. Benzodiazepines enhance the receptor affinity for GABA by decreasing the decay of spontaneous miniature inhibitory postsynaptic currents (mIPSC).
Analgesic
Sedative actions of benzodiazepines limit their usefulness as analgesic agents and they are therefore generally not considered to be appropriate. This limitation can be bypassed by intrathecal administration. GABAA receptors in the periaqueductal gray are pro-nociceptive at supraspinal sites while GABAA that are found in the spinal cord are anti-hyperalgesic. Spinal α2 and α3 containing GABAA receptors are responsible for the anti-hyperalgesic action of intrathecal diazepam. This was shown when the anti-hyperalgesic action was reduced when administered in α2 and α3 mice in inflammatory pain and in neuropathic pain. Additionally, studies in α5 mice showed that the spinal α5-containing GABAA receptor has a minor role in inflammatory pain. An α2, α3 and/or α5 selective positive allosteric agonist, like L-838,427 for example, might be useful as an analgesic drug against inflammatory or neuropathic pain.
Schizophrenia
Benzodiazepines are used as a supporting treatment in patients with schizophrenia.
Depression
GABA ergic hypothesis for depression has been proposed. This hypothesis places the GABA system in a central role in the pathophysiology of depression and in addition to that clinical studies have shown that alprazolam and adinazolam have antidepressant activities in patients with major depressive disorder. Unfortunately we do not know which receptor subtype is responsible for the antidepressant activities. Studies in y2 knockout mice have shown that they display increased anxiety and depressive-like symptoms in despair based tests. The mice also had increased corticosterone concentration which is a symptom in major depression in humans. The y2 subunit is associated with α1-α6 subunits, which are all known α subunits, so these studies do not show which of the α subunits are related to the depressive like symptoms. Other studies with α2 knockout mice have displayed increased anxiety and depression-like symptoms in conflict based feeding tests and the fact that anxiety and depression are often linked seems to indicate that the α2 subunit might be a valid target for a GABAA antidepressant.
Stroke
Preclinical studies have shown that benzodiazepines can be effective in reducing the effect of strokes for up to three days after the drug has been administered.
Neurosteroids
Neurosteroids can act as allosteric modulators of neurotransmitter receptors, such as the GABAA, NMDA, and sigma receptors. The neurosteroid Progesterone (PROG) that activates progesterone receptors expressed in peripheral and central glial cells. Additionally it has been surmised that the 3α-hydroxy ring A-reduced pregnane steroids allopregnanolone and tetrahydrodeoxycorticosterone increase the GABA-mediated chloride currents while pregnenolone sulfate and dehydroepiandrosterone (DHEA) sulfate on the other hand display antagonistic properties at the GABAA receptors.
Barbituric acid
Barbituric acid is the parent compound of barbiturate drugs although barbituric acid itself is not pharmacologically active. Barbiturates were synthesized in 1864 by Adolf von Baeyer by combining urea and malonic acid (Figure 5). A synthesis process was later developed and perfected by French chemist Edouard Grimaux in 1879, making possible the subsequent widespread development of barbiturate derivatives. Malonic acid was later replaced by diethyl malonate, as using the ester avoids the need to deal with the acidity of the carboxylic acid and its unreactive carboxylate (see figure 6). Barbituric acid can form a large variety of barbiturate drugs by using the Knoevenagel condensation reaction.
Benzodiazepines
The structure that the first benzodiazepine is based on was discovered by Leo H. Sternbach. He thought the compound had a heptoxdiazine structure (Figure 7) but it was later determined to be a quinazoline-3-oxide. Possible drug candidates were then synthesized from that compound and screened for activity. One of these compounds was active, chlordiazepoxide. It was marketed in 1960 and became the first benzodiazepine drug.
Biosynthesis of neurosteroids
Neurosteroids are synthesized in the central nervous system (CNS) and the peripheral nervous system (PNS) from cholesterol and steroidal precursors that are imported from the peripheral sources. These sources include 3β-hydroxy-Δ5 derivatives, such as pregnenolone (PREG) and dehydroepiandrosterone (DHEA), their sulfates, and reduced metabolites such as the tetrahydro derivative of progesterone 3α-hydroxy-5α-pregnane-20-one (3α,5α-THPROG). After the local synthesis or from metabolism of adrenal of gonadal steroids many neurosteroids accumulate in the brain.
Barbiturates
Barbiturates have special uses and are organized into 4 classes: ultrashort-, short-, intermediate- and long-acting. Empirically SARs of barbiturants are based on thousands of (animal) tested compounds.They have shown that R and R´ may not be H in position 5 (see figure 8). Also, position 5 confer sedative-hypnotic properties. Generally alkyl branching in position 5 means less lipid solubility and less activity. Unsaturation show less activity in position 5 and alicyclic and aromatic rings show less potency. Polar substiuents (-NH2, -OH, -COOH) will decrease lipid solubility but it will also eliminate activity. R´´ in position 1 is usually, H but CH3 in that position yields less lipid solubility and duration. Exchanging S for O atom in position 2 produces thiobarbiturates, which are more lipid-soluble than the oxybarbiturates. In general, the more lipid-soluble the barbiturate, the more rapid its onset, the shorter its duration and the greater the degree of hypnotic activity. Barbiturates showed some hydrolytic problems in regard to formulation of liquid dosage forms. The difficulty is -OH catalyzed degration of the ureide rings but that can be fixed if the pH is 6 in the formulation. S(-) form of barbiturate have shown more depressant activity while the R(+) isomers have an excitatory effect.
Benzodiazepines
According to research performed by Maddalena et al., using artificial neural networks, position 7 has the most effect on receptor affinity. When the active group in position 7 is made more lipophilic and the electronic charge is increased the receptor affinity increases. In the same study position 2´ was found to be the second-most important in affecting the affinity, but the group in that position needs to be electrophilic to have an effect. Positions 3, 6’ and 8 are of less importance. Changes to 6, 8, 9 or 4´ decrease activity. If the group in position 1 is changed to N-alkyl, haloalkyl, alkynyl and small cycle or aminoalkyl the activity is increased. Position 3 hydroxylation can cause rapid conjugation and decrease duration and potency, which can be clinically useful.
Neurosteroids
In the mid 1980s, the neuroactive steroids 3α,5α-tetrahydroprogesterone or allopregnanolone (3α,5α-THP) and 3α,5α-tetrahydrodeoxycorticosterone (3α,5α-THDOC) were shown to modulate neuronal excitability via their interaction with GABAA receptors. The steroids 3α,5α-THP and 3α,5α-THDOC were able to enhance the GABA-elicited Cl− current. In addition, these steroids might enhance the binding of muscimol and benzodiazepines to GABAA receptors. Structure- activity studies (SAR) showed that the 3alpha-OH group is essential for the anesthetic actions of these steroids, they also have an optimally-placed hydrogen bond accepting group on the β face of the steroid at the C-17 position. The four steroid rings form a rigid framework for positioning these hydrogen groups in three-dimensional space. Analogues 5 and 6 (Figure 10) are weak modulators of GABAA receptor function because the flexible side chains in these analogues do not have the conformations required for high biological activity.