
 | ||
Cancer stem cells (CSCs) are cancer cells (found within tumors or hematological cancers) that possess characteristics associated with normal stem cells, specifically the ability to give rise to all cell types found in a particular cancer sample. CSCs are therefore tumorigenic (tumor-forming), perhaps in contrast to other non-tumorigenic cancer cells. CSCs may generate tumors through the stem cell processes of self-renewal and differentiation into multiple cell types. Such cells are hypothesized to persist in tumors as a distinct population and cause relapse and metastasis by giving rise to new tumors. Therefore, development of specific therapies targeted at CSCs holds hope for improvement of survival and quality of life of cancer patients, especially for patients with metastatic disease.
Contents
- Tumor propagation models
- The Cancer Stem Cell Model
- Stochastic Model
- Tying CSC and Stochastic Models Together
- Debate
- Evidence
- Mechanistic and mathematical models
- Origin
- Stem cell mutation
- Adult stem cells
- De differentiation
- Hierarchy
- Identification
- Heterogeneity markers
- Metastasis
- Epithelial mesenchymal transition
- Two phase expression pattern
- Implications
- Treatment
- Targeting
- Pathways
- BMI 1
- Notch
- Sonic hedgehog and Wnt
- References
Existing cancer treatments have mostly been developed based on animal models, where therapies able to promote tumor shrinkage were deemed effective. However, animals do not provide a complete model of human disease. In particular, in mice, whose life spans do not exceed two years, tumor relapse is difficult to study.
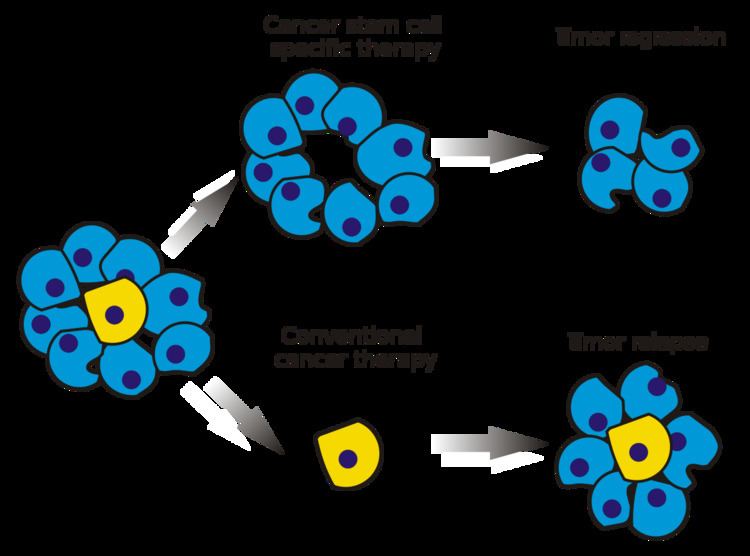
The efficacy of cancer treatments is, in the initial stages of testing, often measured by the ablation fraction of tumor mass (fractional kill). As CSCs form a small proportion of the tumor, this may not necessarily select for drugs that act specifically on the stem cells. The theory suggests that conventional chemotherapies kill differentiated or differentiating cells, which form the bulk of the tumor but do not generate new cells. A population of CSCs, which gave rise to it, could remain untouched and cause relapse.
Cancer stem cells were first identified by John Dick in acute myeloid leukemia in the late 1990s. Since the early 2000s they have been an intense cancer research focus.
Tumor propagation models
In different tumor subtypes, cells within the tumor population exhibit functional heterogeneity and tumors are formed from cells with various proliferative and differentiation capacities. This functional heterogeneity among cancer cells has led to the creation of multiple propagation models to account for heterogeneity and differences in tumor-regenerative capacity: the cancer stem cell (CSC) and stochastic model.
The Cancer Stem Cell Model
The Cancer Stem Cell Model, also known as the Hierarchical Model proposes that tumors are hierarchically organized (CSCs lying at the apex (Fig. 3).) Within the cancer population of the tumors there are cancer stem cells (CSC) that are tumorigenic cells and are biologically distinct from other subpopulations They have two defining features: their long-term ability to self-renew and their capacity to differentiate into progeny that is non-tumorigenic but still contributes to the growth of the tumor. This model suggests that only certain subpopulations of cancer stem cells have the ability to drive the progression of cancer, meaning that there are specific (intrinsic) characteristics that can be identified and then targeted to destroy a tumor long-term without the need to battle the whole tumor
Stochastic Model
In order for a cell to become cancerous it must undergo a significant number of alterations to its DNA sequence. This cell model suggests these mutations could occur to any cell in the body resulting in a cancer. Essentially this theory proposes that all cells have the ability to be tumorigenic making all tumor cells equipotent with the ability to self-renew or differentiate, leading to tumor heterogeneity while others can differentiate into non-CSCs The cell's potential can be influenced by unpredicted genetic or epigenetic factors, resulting in phenotypically diverse cells in both the tumorigenic and non-tumorigenic cells that compose the tumor.
These mutations could progressively accumulate and enhance the resistance and fitness of cells that allow them to outcompete other tumor cells, better known as the somatic evolution model. The clonal evolution model, which occurs in both the CSC model and stochastic model, postulates that mutant tumor cells with a growth advantage outproliferate others. Cells in the dominant population have a similar potential for initiating tumor growth (Fig. 4).
These two models are not mutually exclusive, as CSCs themselves undergo clonal evolution. Thus, the secondary more dominant CSCs may emerge, if a mutation confers more aggressive properties (Fig. 5).
Tying CSC and Stochastic Models Together
A study in 2014 argues the gap between these two controversial models can be bridged by providing an alternative explanation of tumor heterogeneity. They demonstrate a model that includes aspects of both the Stochastic and CSC models. They examined cancer stem cell plasticity in which cancer stem cells can transition between non-cancer stem cells (Non-CSC) and CSC via in situ supporting a more Stochastic model. But the existence of both biologically distinct non-CSC and CSC populations supports a more CSC model, proposing that both models may play a vital role in tumor heterogeneity.
Debate
The existence of CSCs is under debate, because many studies found no cells with their specific characteristics. Cancer cells must be capable of continuous proliferation and self-renewal to retain the many mutations required for carcinogenesis and to sustain the growth of a tumor, since differentiated cells (constrained by the Hayflick Limit) cannot divide indefinitely. If most tumor cells are endowed with stem cell properties, targeting tumor size directly is a valid strategy. If they are a small minority, targeting them may be more effective. Another debate is over the origin of CSCs - whether from disregulation of normal stem cells or from a more specialized population that acquired the ability to self-renew (which is related to the issue of stem cell plasticity). Confounding this debate is the discovery that many cancer cells demonstrate a Phenotypic plasticity under therapeutic challenge, altering their transcriptomes to a more stem-like state to escape destruction.
Evidence
The first conclusive evidence for CSCs came in 1997. Bonnet and Dick isolated a subpopulation of leukemia cells that expressed surface marker CD34, but not CD38. The authors established that the CD34+/CD38− subpopulation is capable of initiating tumors in NOD/SCID mice that were histologically similar to the donor. The first evidence of a solid tumor cancer stem-like cell followed in 2002 with the discovery of a clonogenic, sphere-forming cell isolated and characterized from human brain gliomas. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro.
In cancer research experiments, tumor cells are sometimes injected into an experimental animal to establish a tumor. Disease progression is then followed in time and novel drugs can be tested for their efficacy. Tumor formation requires thousands or tens of thousands of cells to be introduced. Classically, this was explained by poor methodology (i.e., the tumor cells lose their viability during transfer) or the critical importance of the microenvironment, the particular biochemical surroundings of the injected cells. Supporters of the CSC paradigm argue that only a small fraction of the injected cells, the CSCs, have the potential to generate a tumor. In human acute myeloid leukemia the frequency of these cells is less than 1 in 10,000.
Further evidence comes from histology. Many tumors are heterogeneous and contain multiple cell types native to the host organ. Heterogeneity is commonly retained by tumor metastases. This suggests that the cell that produced them had the capacity to generate multiple cell types, a classical hallmark of stem cells.
The existence of leukemia stem cells prompted research into other cancers. CSCs have recently been identified in several solid tumors, including:
Mechanistic and mathematical models
Once the pathways to cancer are hypothesized, it is possible to develop predictive mathematical models, e.g., based on the cell compartment method. For instance, the growths of abnormal cells can be denoted with specific mutation probabilities. Such a model predicted that repeated insult to mature cells increases the formation of abnormal progeny and the risk of cancer. The clinical efficacy of such models remains unestablished.
Origin
The origin of CSCs is an active research area. The answer may depend on the tumor type and phenotype. So far the hypothesis that tumors originate from a single "cell of origin" has not been demonstrated using the cancer stem cell model. This is because cancer stem cells are not present in end-stage tumors.
Origin hypotheses include mutants in developing stem or progenitor cells, mutants in adult stem cells or adult progenitor cells and mutant, differentiated cells that acquire stem-like attributes. These theories often focus on a tumor's "cell of origin".
Stem cell mutation
The "mutation in stem cell niche populations during development" hypothesis claims that these developing stem populations are mutated and then reproduce so that the mutation is shared by many descendants. These daughter cells are much closer to becoming tumors and their numbers increase the chance of a cancerous mutation.
Adult stem cells
Another theory associates adult stem cells (ASC) with tumor formation. This is most often associated with tissues with a high rate of cell turnover (such as the skin or gut). In these tissues, ASCs are candidates because of their frequent cell divisions (compared to most ASCs) in conjunction with the long lifespan of ASCs. This combination creates the ideal set of circumstances for mutations to accumulate: mutation accumulation is the primary factor that drives cancer initiation. Evidence shows that the association represents an actual phenomenon, although specific cancers have been linked to a specific cause.
De-differentiation
De-differentiation of mutated cells may create stem cell-like characteristics, suggesting that any cell might become a cancer stem cell. In other words, a fully differentiated cell undergoes mutations or extracellular signals that drive it back to a stem-like state. This concept has been demonstrated most recently in Prostate cancer models, whereby cells undergoing androgen deprivation therapy appear to transiently alter their transcriptome to that of a neural crest stem-like cell, with the invasive and multipotent properties of this class of stem-like cells.
Hierarchy
The concept of tumor hierarchy claims that a tumor is a heterogeneous population of mutant cells, all of which share some mutations, but vary in specific phenotype. A tumor hosts several types of stem cells, one optimal to the specific environment and other less successful lines. These secondary lines may be more successful in other environments, allowing the tumor to adapt, including adaptation to therapeutic intervention. If correct, this concept impacts cancer stem cell-specific treatment regimes. Such a hierarchy would complicate attempts to pinpoint the origin.
Identification
CSCs, now reported in most human tumors, are commonly identified and enriched using strategies for identifying normal stem cells that are similar across studies. These procedures include fluorescence-activated cell sorting (FACS), with antibodies directed at cell-surface markers and functional approaches including side population assay or Aldefluor assay. The CSC-enriched result is then implanted, at various doses, in immune-deficient mice to assess its tumor development capacity. This in vivo assay is called a limiting dilution assay. The tumor cell subsets that can initiate tumor development at low cell numbers are further tested for self-renewal capacity in serial tumor studies.
CSC can also be identified by efflux of incorporated Hoechst dyes via multidrug resistance (MDR) and ATP-binding cassette (ABC) Transporters.
Another approach is sphere-forming assays. Many normal stem cells such as hematopoietics or stem cells from tissues, under special culture conditions, form three-dimensional spheres that can differentiate. As with normal stem cells, the CSCs isolated from brain or prostate tumors also have the ability to form anchor-independent spheres.
Heterogeneity (markers)
CSCs have been identified in various solid tumors. Markers specific for normal stem cells are commonly used for isolating CSCs from solid and hematological tumors. Cell surface markers have proved useful for isolation of CSC-enriched populations including CD133 (also known as PROM1), CD44, CD24, EpCAM (epithelial cell adhesion molecule, also known as epithelial specific antigen, ESA), THY1, ATP-binding cassette B5 (ABCB5), and CD200.
CD133 (prominin 1) is a five-transmembrane domain glycoprotein expressed on CD34+ stem and progenitor cells, in endothelial precursors and fetal neural stem cells. It has been detected using its glycosylated epitope known as AC133. However, the use of this marker has proven controversial as it is an inconsistent marker of CSCs.
EpCAM (epithelial cell adhesion molecule, ESA, TROP1) is hemophilic Ca2+-independent cell adhesion molecule expressed on the basolateral surface of most epithelial cells.
CD90 (THY1) is a glycosylphosphatidylinositol glycoprotein anchored in the plasma membrane and involved in signal transduction. It may also mediate adhesion between thymocytes and thymic stroma.
CD44 (PGP1) is an adhesion molecule that has pleiotropic roles in cell signaling, migration and homing. It has multiple isoforms, including CD44H, which exhibits high affinity for hyaluronate and CD44V which has metastatic properties. Like CD133, the use of CD44 as a CSC marker has proven controversial, as it is also an inconsistent marker of CSCs.
CD24 (HSA) is a glycosylated glycosylphosphatidylinositol-anchored adhesion molecule, which has co-stimulatory role in B and T cells.
CD200 (OX-2) is a type 1 membrane glycoprotein, which delivers an inhibitory signal to immune cells including T cells, natural killer cells and macrophages.
ALDH is a ubiquitous aldehyde dehydrogenase family of enzymes, which catalyzes the oxidation of aromatic aldehydes to carboxyl acids. For instance, it has a role in conversion of retinol to retinoic acid, which is essential for survival.
The first solid malignancy from which CSCs were isolated and identified was breast cancer and they are the most intensely studied. Breast CSCs have been enriched in CD44+CD24−/low, SP and ALDH+ subpopulations. Breast CSCs are apparently phenotypically diverse. CSC marker expression in breast cancer cells is apparently heterogeneous and breast CSC populations vary across tumors. Both CD44+CD24− and CD44+CD24+ cell populations are tumor initiating cells; however, CSC are most highly enriched using the marker profile CD44+CD49fhiCD133/2hi.
CSCs have been reported in many brain tumors. Stem-like tumor cells have been identified using cell surface markers including CD133, SSEA-1 (stage-specific embryonic antigen-1), EGFR and CD44. The use of CD133 for identification of brain tumor stem-like cells may be problematic because tumorigenic cells are found in both CD133+ and CD133− cells in some gliomas and some CD133+ brain tumor cells may not possess tumor-initiating capacity.
CSCs were reported in human colon cancer. For their identification, cell surface markers such as CD133, CD44 and ABCB5, functional analysis including clonal analysis and Aldefluor assay were used. Using CD133 as a positive marker for colon CSCs generated conflicting results. The AC133 epitope, but not the CD133 protein, is specifically expressed in colon CSCs and its expression is lost upon differentiation. In addition, CD44+ colon cancer cells and additional sub-fractionation of CD44+EpCAM+ cell population with CD166 enhance the success of tumor engraftments.
Multiple CSCs have been reported in prostate, lung and many other organs, including liver, pancreas, kidney or ovary. In prostate cancer, the tumor-initiating cells have been identified in CD166+α2β1+, CD44+, CD44+α2β1+, TRA-1-60+CD151+CD166+ or ALDH+ cell populations. Putative markers for lung CSCs have been reported, including CD133+, ALDH+, CD44+ and oncofetal protein 5T4+.
Metastasis
Metastasis is the major cause of tumor lethality. However, not every tumor cell can metastasize. This potential depends on factors that determine growth, angiogenesis, invasion and other basic processes.
Epithelial-mesenchymal transition
In epithelial tumors, the epithelial-mesenchymal transition (EMT) is considered to be a crucial event. EMT and the reverse transition from mesenchymal to an epithelial phenotype (MET) are involved in embryonic development, which involves disruption of epithelial cell homeostasis and the acquisition of a migratory mesenchymal phenotype. EMT appears to be controlled by canonical pathways such as WNT and transforming growth factor β.
EMT's important feature is the loss of membrane E-cadherin in adherens junctions, where β-catenin may play a significant role. Translocation of β-catenin from adherens junctions to the nucleus may lead to a loss of E-cadherin and subsequently to EMT. Nuclear β-catenin apparently can directly, transcriptionally activate EMT-associated target genes, such as the E-cadherin gene repressor SLUG (also known as SNAI2). Mechanical properties of the tumor microenvironment, such as hypoxia, can contribute to CSC survival and metastatic potential through stabilization of hypoxia inducible factors through interactions with ROS (reactive oxygen species).
Tumor cells undergoing an EMT may be precursors for metastatic cancer cells, or even metastatic CSCs. In the invasive edge of pancreatic carcinoma, a subset of CD133+CXCR4+ (receptor for CXCL12 chemokine also known as a SDF1 ligand) cells was defined. These cells exhibited significantly stronger migratory activity than their counterpart CD133+CXCR4− cells, but both showed similar tumor development capacity. Moreover, inhibition of the CXCR4 receptor reduced metastatic potential without altering tumorigenic capacity.
Two-phase expression pattern
In breast cancer CD44+CD24−/low cells are detectable in metastatic pleural effusions. By contrast, an increased number of CD24+ cells have been identified in distant metastases in breast cancer patients. It is possible that CD44+CD24−/low cells initially metastasize and in the new site change their phenotype and undergo limited differentiation. The two-phase expression pattern hypothesis proposes two forms of cancer stem cells - stationary (SCS) and mobile (MCS). SCS are embedded in tissue and persist in differentiated areas throughout tumor progression. MCS are located at the tumor-host interface. These cells are apparently derived from SCS through the acquisition of transient EMT (Figure 7).
Implications
CSCs have implications for cancer therapy, including for disease identification, selective drug targets, prevention of metastasis and intervention strategies.
Treatment
Somatic stem cells are naturally resistant to chemotherapeutic agents. They produce various pumps (such as MDR) that pump out drugs and DNA repair proteins. They have a slow rate of cell turnover (chemotherapeutic agents naturally target rapidly replicating cells). CSCs that develop from normal stem cells may also produce these proteins, which could increase their resistance towards chemotherapy. The surviving CSCs then repopulate the tumor, causing a relapse.
Targeting
Selectively targeting CSCs may allow treatment of aggressive, non-resectable tumors, as well as prevent metastasis and relapse. The hypothesis suggests that upon CSC elimination, cancer could regress due to differentiation and/or cell death. The fraction of tumor cells that are CSCs and therefore need to be eliminated is unclear.
Studies looked for specific markers and for proteomic and genomic tumor signatures that distinguish CSCs from others. In 2009, scientists identified the compound salinomycin, which selectively reduces the proportion of breast CSCs in mice by more than 100-fold relative to Paclitaxel, a commonly used chemotherapeutic agent. Some types of cancer cells can survive treatment with salinomycin through autophagy, whereby cells use acidic organelles such as lysosomes to degrade and recycle certain types of proteins. The use of autophagy inhibitors can kill cancer stem cells that survive by autophagy.
The cell surface receptor interleukin-3 receptor-alpha (CD123) is overexpressed on CD34+CD38- leukemic stem cells (LSCs) in acute myelogenous leukemia (AML) but not on normal CD34+CD38- bone marrow cells. Treating AML-engrafted NOD/SCID mice with a CD123-specific monoclonal antibody impaired LSCs homing to the bone marrow and reduced overall AML cell repopulation including the proportion of LSCs in secondary mouse recipients.
A 2015 study packaged nanoparticles with miR-34a and ammonium bicarbonate and delivered them to prostate CSCs in a mouse model. Then they irradiated the area with near-infrared laser light. This caused the nanoparticles to swell three times or more in size bursting the endosomes and dispersing the RNA in the cell. miR-34a can lower the levels of CD44.
Pathways
The design of new drugs for targeting CSCs requires understanding the cellular mechanisms that regulate cell proliferation. The first advances in this area were made with hematopoietic stem cells (HSCs) and their transformed counterparts in leukemia, the disease for which the origin of CSCs is best understood. Stem cells of many organs share the same cellular pathways as leukemia-derived HSCs.
A normal stem cell may be transformed into a CSC through disregulation of the proliferation and differentiation pathways controlling it or by inducing oncoprotein activity.
BMI-1
The Polycomb group transcriptional repressor Bmi-1 was discovered as a common oncogene activated in lymphoma and later shown to regulate HSCs. The role of Bmi-1 has been illustrated in neural stem cells. The pathway appears to be active in CSCs of pediatric brain tumors.
Notch
The Notch pathway plays a role in controlling stem cell proliferation for several cell types including hematopoietic, neural and mammary SCs. Components of this pathway have been proposed to act as oncogenes in mammary and other tumors.
A branch of the Notch signaling pathway that involves the transcription factor Hes3 regulates a number of cultured cells with CSC characteristics obtained from glioblastoma patients.
Sonic hedgehog and Wnt
These developmental pathways are SC regulators. Both Sonic hedgehog (SHH) and Wnt pathways are commonly hyperactivated in tumors and are necessary to sustain tumor growth. However, the Gli transcription factors that are regulated by SHH take their name from gliomas, where they are highly expressed. A degree of crosstalk exists between the two pathways and they are commonly activated together. By contrast, in colon cancer hedgehog signalling appears to antagonise Wnt.
Sonic hedgehog blockers are available, such as cyclopamine. A water-soluble cyclopamine may be more effective in cancer treatment. DMAPT, a water-soluble derivative of parthenolide, induces oxidative stress and inhibits NF-κB signaling for AML (leukemia) and possibly myeloma and prostate cancer. Telomerase is a study subject in CSC physiology. GRN163L (Imetelstat) was recently started in trials to target myeloma stem cells.
Wnt signaling can become independent of regular stimuli, through mutations in downstream oncogenes and tumor suppressor genes that become permanently activated even though the normal receptor has not received a signal. β-catenin binds to transcription factors such as the protein TCF4 and in combination the molecules activate the necessary genes. LF3 strongly inhibits this binding in vitro, in cell lines and reduced tumor growth in mouse models. It prevented replication and reduced their ability to migrate, all without affecting healthy cells. No cancer stem cells remained after treatment. The discovery was the product of "rational drug design", involving AlphaScreens and ELISA technologies.