
 | ||
Autophagy (or autophagocytosis) (from the Ancient Greek αὐτόφαγος autóphagos, meaning "self-devouring" and κύτος kýtos, meaning "hollow") is the natural, regulated, destructive mechanism of the cell that disassembles unnecessary or dysfunctional components.
Contents
- History
- Process and pathways
- Molecular biology
- Nutrient starvation
- Xenophagy
- Autophagy and infection
- Repair mechanism
- Programmed cell death
- Autophagy and caloric restriction
- Autophagy and exercise
- Autophagy and osteoarthritis
- Autophagy and cancer
- Tumor suppressor
- Tumor cell survival
- Mechanism of cell death
- Therapeutic target
- Autophagy and Parkinson disease
- References
Autophagy allows the orderly degradation and recycling of cellular components. In macroautophagy, targeted cytoplasmic constituents are isolated from the rest of the cell within a double-membraned vesicle known as an autophagosome. The autophagosome eventually fuses with lysosomes and the contents are degraded and recycled. Two additional forms of autophagy are also commonly described: microautophagy and chaperone-mediated autophagy (CMA). In disease, autophagy has been seen as an adaptive response to stress, which promotes survival, whereas in other cases it appears to promote cell death and morbidity. In the extreme case of starvation, the breakdown of cellular components promotes cellular survival by maintaining cellular energy levels.
The name "autophagy" was coined by Belgian biochemist Christian de Duve in 1963. The identification of autophagy-related genes in yeast in the 1990s let researchers figure out the mechanisms of autophagy, and led to the award of the 2016 Nobel Prize in Physiology or Medicine to Japanese autophagy researcher Yoshinori Ohsumi.
History
Autophagy was first observed by Keith R. Porter and his student Thomas Ashford at the Rockefeller Institute. In January 1962 they reported an increased number of lysosomes in rat liver cells after addition of glucagon, and that some displaced lysosomes towards the centre of the cell contained other cell organelles such as mitochondria. They called this autolysis after Christian de Duve and Alex B. Novikoff. However Porter and Ashford wrongly interpreted their data as lysosome formation (ignoring the pre-existing organelles). Lysosomes could not be cell organelles, but part of cytoplasm such as mitochondria, and that hydrolytic enzymes were produced by microbodies. In 1963 researchers published a detailed ultrastructural description of "focal cytoplasmic degradation," which referenced a 1955 German study of injury-induced sequestration. The study recognized three continuous stages of maturation of the sequestered cytoplasm to lysosomes, and that the process was not limited to injury states that functioned under physiological conditions for "reutilization of cellular materials," and the "disposal of organelles" during differentiation. Inspired by this discovery, de Duve christened the phenomena "autophagy". Unlike Porter and Ashford, de Duve conceived the term as a part of lysosomal function while describing the role of glucagon as a major inducer of cell degradation in the liver. With his student Peter, he established that lysosomes are responsible for glucagon-induced autophagy. This was the first time the fact that lysosomes were established as the sites of intracellular autophagy.
A new era of autophagy research began in 1990s when several groups of scientists independently discovered autophagy-related genes using the budding yeast. Notably, Yoshinori Ohsumi and Michael Thumm examined starvation-induced non-selective autophagy; in the meantime, Daniel J Klionsky discovered the Cytoplasm-to-Vacuole Targeting (CVT) pathway, which is a form of selective autophagy. They soon found that they were in fact looking at essentially the same pathway, just from different angles. Initially, the genes discovered by these and other yeast groups were given different names (APG, AUT, CVT, GSA, PAG, PAZ, and PDD). A unified nomenclature was advocated in 2003 by the yeast researchers to use ATG to denote autophagy genes. The 2016 Nobel Prize in Physiology or Medicine was awarded to Yoshinori Ohsumi. Dr. Ohsumi’s contribution to autophagy research is well recognized. However, some has pointed out that the award could have been more inclusive.
The field of autophagy research experienced accelerated growth at the turn of the century. Knowledge of ATG genes provided scientists more convenient tools to dissect functions of autophagy in human health and disease. In 1999, a landmark discovery connecting autophagy with cancer was published by Beth Levine’s group. To this date, relationship between cancer and autophagy continues to be a main theme of autophagy research. The roles of autophagy in neurodegeneration and immune defense also received considerable attention. In 2003, the first Gordon conference on autophagy was held at Waterville. In 2005, Daniel J Klionsky launched “Autophagy”, a scientific journal dedicated to this field. The first Keystone Symposia on autophagy was held in 2007 at Monterey.
Process and pathways
There are three pathways of autophagy and these are mediated by the autophagy-related genes and their associated enzymes.
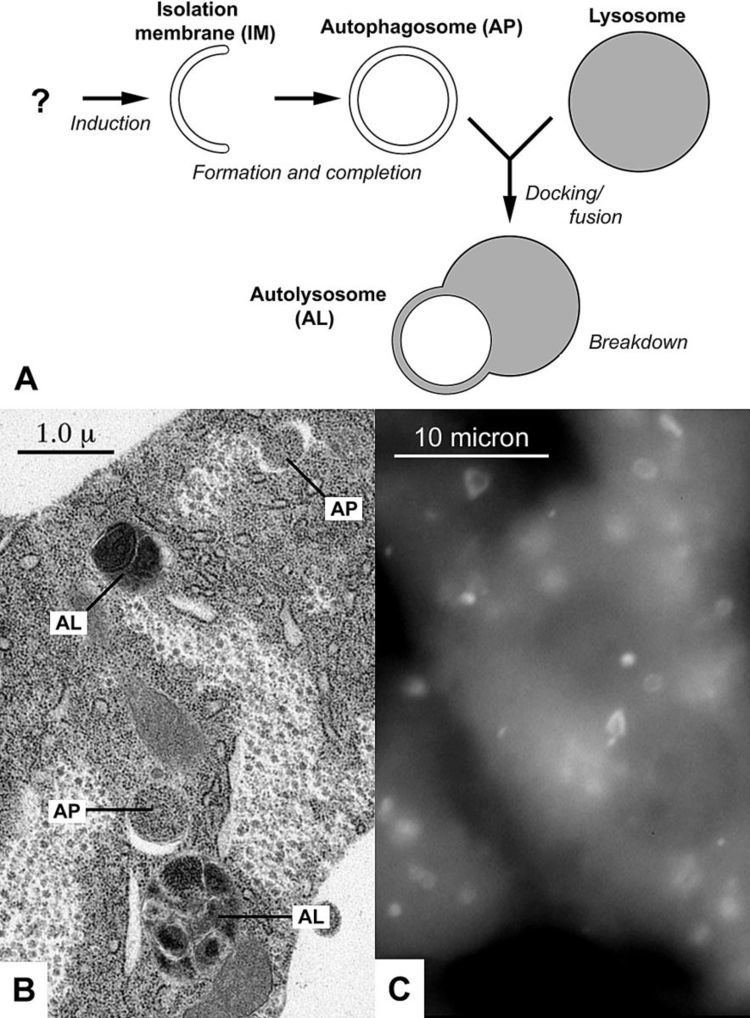
Macroautophagy is the main pathway, used primarily to eradicate damaged cell organelles or unused proteins. This involves the formation of a double membrane known as an autophagosome, around the organelle marked for destruction. The autophagosome then travels through the cytoplasm of the cell to a lysosome, and the two organelles fuse. Within the lysosome, the contents of the autophagosome are degraded via acidic lysosomal hydrolases.
Microautophagy, on the other hand, involves the direct engulfment of cytoplasmic material into the lysosome. This occurs by invagination, meaning the inward folding of the lysosomal membrane, or cellular protrusion.
Chaperone-mediated autophagy, or CMA, is a very complex and specific pathway, which involves the recognition by the hsc70-containing complex. This means that a protein must contain the recognition site for this hsc70 complex which will allow it to bind to this chaperone, forming the CMA- substrate/chaperone complex. This complex then moves to the lysosomal membrane-bound protein that will recognise and bind with the CMA receptor, allowing it to enter the cell. Upon recognition, the substrate protein gets unfolded and it is translocated across the lysosome membrane with the assistance of the lysosomal hsc70 chaperone. CMA is significantly different from other types of autophagy because it translocates protein material in a one by one manner, and it is extremely selective about what material crosses the lysosomal barrier.
Molecular biology
Autophagy is executed by autophagy-related (Atg) genes. The first autophagy genes were identified by genetic screens conducted in the budding yeast Saccharomyces cerevisiae. Following their identification those genes were functionally characterized and their orthologs in a variety of different organisms were identified and studied.
In mammals, amino acid sensing and additional signals such as growth factors and reactive oxygen species regulate the activity of the protein kinases mTOR and AMPK. These two kinases regulate autophagy through inhibitory phosphorylation of the Unc-51-like kinases ULK1 and ULK2 (mammalian homologues of Atg1). Induction of autophagy results in the dephosphorylation and activation of the ULK kinases. ULK is part of a protein complex containing Atg13, Atg101 and FIP200. ULK phosphorylates and activates Beclin-1 (mammalian homologue of Atg6), which is also part of a protein complex. The autophagy-inducible Beclin-1 complex contains the proteins p150, Atg14L and the class III phosphatidylinositol 3-phosphate kinase (PI(3)K) VPS34. The active ULK and Beclin-1 complexes re-localize to the site of autophagosome initiation, the phagophore, where they both contribute to the activation of downstream autophagy components.
Once active, VPS34 phosphorylates the lipid phosphatidylinositol to generate phosphatidylinositol 3-phosphate (PtdIns(3)P) on the surface of the phagophore. The generated PtdIns(3)P is used as a docking point for proteins harboring a PtdIns(3)P binding motif. WIPI2, a PtdIns(3)P binding protein of the WIPI (WD-repeat protein interacting with phosphoinositides) protein family, was recently shown to physically bind Atg16L1. Atg16L1 is a member of an E3-like protein complex involved in one of two ubiquitin-like conjugation systems essential for autophagosome formation. Its binding by WIPI2 recruits it to the phagophore and mediates its activity.
The first of the two ubiquitin-like conjugation systems involved in autophagy covalently binds the ubiquitin-like protein Atg12 to Atg5. The resulting conjugate protein then binds Atg16L1 to form an E3-like complex which functions as part of the second ubiquitin-like conjugation system. This complex binds and activates Atg3, which covalently attaches mammalian homologues of the ubiquitin-like yeast protein Atg8 (LC3A-C, GATE16, and GABARAPL1-3), the most studied being LC3 proteins, to the lipid phosphatidylethanolamine (PE) on the surface of autophagosomes. Lipidated LC3 contributes to the closure of autophagosomes, and enables the docking of specific cargos and adaptor proteins such as Sequestosome-1/p62. The completed autophagosome then fuses with a lysosome through the actions of multiple proteins, including SNAREs and UVRAG. Following the fusion LC3 is retained on the vesicle's inner side and degraded along with the cargo, while the LC3 molecules attached to the outer side are cleaved off by Atg4 and recycled. The contents of the autolysosome are subsequently degraded and their building blocks are released from the vesicle through the action of permeases.
Nutrient starvation
Autophagy has roles in various cellular functions. One particular example is in yeasts, where the nutrient starvation induces a high level of autophagy. This allows unneeded proteins to be degraded and the amino acids recycled for the synthesis of proteins that are essential for survival. In higher eukaryotes, autophagy is induced in response to the nutrient depletion that occurs in animals at birth after severing off the trans-placental food supply, as well as that of nutrient starved cultured cells and tissues. Mutant yeast cells that have a reduced autophagic capability rapidly perish in nutrition-deficient conditions. Studies on the apg mutants suggest that autophagy via autophagic bodies is indispensable for protein degradation in the vacuoles under starvation conditions, and that at least 15 APG genes are involved in autophagy in yeast. A gene known as ATG7 has been implicated in nutrient-mediated autophagy, as mice studies have shown that starvation-induced autophagy was impaired in atg7-deficient mice.
Xenophagy
In microbiology, xenophagy is the autophagic degradation of infectious particles. Cellular autophagic machinery also play an important role in innate immunity. Intracellular pathogens, such as Mycobacterium tuberculosis (the bacterium which is responsible for tuberculosis) are targeted for degradation by the same cellular machinery and regulatory mechanisms that target host mitochondria for degradation. Incidentally, this is further evidence for the endosymbiotic hypothesis. This process generally leads to the destruction of the invasive organism, although some bacteria can block the maturation of phagosomes into degradative organelles called phagolysosomes. Stimulation of autophagy in infected cells can help overcome this phenomenon, restoring pathogen degradation.
Autophagy and infection
Vesicular stomatitis virus is believed to be taken up by the autophagosome from the cytosol and translocated to the endosomes where detection takes place by a member of the PRRs called toll-like receptor 7, detecting single stranded RNA. Following activation of the toll-like receptor, intracellular signaling cascades are initiated, leading to induction of interferon and other antiviral cytokines. A subset of viruses and bacteria subvert the autophagic pathway to promote their own replication. Galectin-8 has recently been identified as an intracellular "danger receptor", able to initiate autophagy against intracellular pathogens. When galectin-8 binds to a damaged vacuole, it recruits autophagy adaptor such as NDP52 leading to the formation of an autophagosome and bacterial degradation.
Repair mechanism
Autophagy degrades damaged organelles, cell membranes and proteins, and the failure of autophagy is thought to be one of the main reasons for the accumulation of cell damage and aging.
Programmed cell death
One of the mechanisms of programmed cell death (PCD) is associated with the appearance of autophagosomes and depends on autophagy proteins. This form of cell death most likely corresponds to a process that has been morphologically defined as autophagic PCD. One question that constantly arises, however, is whether autophagic activity in dying cells is the cause of death or is actually an attempt to prevent it. Morphological and histochemical studies so far did not prove a causative relationship between the autophagic process and cell death. In fact, there have recently been strong arguments that autophagic activity in dying cells might actually be a survival mechanism. Studies of the metamorphosis of insects have shown cells undergoing a form of PCD that appears distinct from other forms; these have been proposed as examples of autophagic cell death. Recent pharmacological and biochemical studies have proposed that survival and lethal autophagy can be distinguished by the type and degree of regulatory signaling during stress particularly after viral infection. Although promising, these findings have not been examined in non-viral systems.
Autophagy and caloric restriction
Research suggests that autophagy is required for the lifespan-prolonging effects of caloric restriction. A 2010 French study of nematodes, mice and flies showed that inhibition of autophagy exposed cells to metabolic stress. Resveratrol and the dietary restriction prolonged the lifespan of normal, autophagy-proficient nematodes, but not of nematodes in which autophagy had been inhibited by knocking out Beclin 1 (a known autophagic modulator).
Autophagy and exercise
Autophagy is essential for basal homeostasis; it is also extremely important in maintaining muscle homeostasis during physical exercise. Autophagy at the molecular level is only partially understood. A study of mice shows that autophagy is important for the ever changing demands of their nutritional and energy needs, particularly through the metabolic pathways of protein catabolism. In a 2012 study conducted by the University of Texas Southwestern Medical Center in Dallas, mutant mice (with a knock-in mutation of BCL2 phosphorylation sites to produce progeny that showed normal levels of basal autophagy yet were deficient in stress-induced autophagy) were tested to challenge this theory. Results showed that when compared to a control group, these mice illustrated a decrease in endurance and an altered glucose metabolism during acute exercise.
Another study demonstrated that skeletal muscle fibres of collagen VI knockout mice showed signs of degeneration due to an insufficiency of autophagy which led to an accumulation of damaged mitochondria and excessive cell death. Exercise-induced autophagy was unsuccessful however; but when autophagy was induced artificially post-exercise, the accumulation of damaged organelles in collagen VI deficient muscle fibres was prevented and cellular homeostasis was maintained. Both studies demonstrate that autophagy induction may contribute to the beneficial metabolic effects of exercise and that it is essential in the maintaining of muscle homeostasis during exercise, particularly in collagen VI fibres.
Work at the Institute for Cell Biology, University of Bonn, showed that a certain type of autophagy, i.e., chaperone-assisted selective autophagy (CASA), is induced in contracting muscles and is required for maintaining the muscle sarcomere under mechanical tension. The CASA chaperone complex recognizes mechanically damaged cytoskeleton components and directs these components through a ubiquitin-dependent autophagic sorting pathway to lysosomes for disposal. This is necessary for maintaining muscle activity.
Autophagy and osteoarthritis
Because autophagy decreases with age and age is a major risk factor for osteoarthritis, the role of autophagy in the development of this disease is suggested. Proteins involved in autophagy are reduced with age in both human and mouse articular cartilage. Mechanical injury to cartilage explants in culture also reduced autophagy proteins. Autophagy is constantly activated in normal cartilage but it is compromised with age and precedes cartilage cell death and structural damage. These results suggest autophagy is a normal protective process (chondroprotection) in the joint.
Autophagy and cancer
Oftentimes, cancer occurs when several different pathways that regulate cell differentiation are disturbed. Autophagy plays an important role in cancer – both in protecting against cancer as well as potentially contributing to the growth of cancer. Autophagy can contribute to cancer by promoting survival of tumor cells that have been starved, or that degrade apoptotic mediators through autophagy: in such cases, use of inhibitors of the late stages of autophagy (such as chloroquine), on the cells that use autophagy to survive, increases the number of cancer cells killed by antineoplastic drugs.
The role of autophagy in cancer is one that has been highly researched and reviewed. There is evidence that emphasizes the role of autophagy both as a tumor suppressor as well as a factor in tumor cell survival. However, recent research has been able to show that autophagy is more likely to be used as a tumor suppressor according to several models.
Tumor suppressor
Several experiments have been done with mice and varying Beclin1, a protein that regulates autophagy. When the Beclin1 gene was altered to be heterozygous (Beclin 1+/-), the mice were found to be tumor prone. However, when Beclin1 was overexpressed, tumor development was inhibited. Care should be exercised when interpreting phenotypes of beclin mutants and attributing the observations to a defect in autophagy, however: Beclin1 is generally required for phosphatidylinositol 3- phosphate production and as such it affects numerous lysosomal and endosomal functions, including endocytosis and endocytic degradation of activated growth factor receptors. In support of the possibility that Beclin1 affects cancer development through an autophagy-independent pathway is the fact that core autophagy factors which are not known to affect other cellular processes and are definitely not known to affect cell proliferation and cell death, such as Atg7 or Atg5, show a much different phenotype when the respective gene is knocked out, which does not include tumor formation. In addition, full knockout of Beclin1 is embryonic lethal whereas knockout of Atg7 or Atg5 is not.
Necrosis and chronic inflammation also has been shown to be limited through autophagy which helps protect against the formation of tumor cells. Thus these experiments show autophagy’s role as a tumor suppressor.
Tumor cell survival
Alternatively, autophagy has also been shown to play a huge role in tumor cell survival. In cancerous cells, autophagy is used as a way to deal with stress on the cell. Once these autophagy related genes were inhibited, cell death was potentiated. The increase in metabolic energy is offset by autophagy functions. These metabolic stresses include hypoxia, nutrient deprivation, and an increase in proliferation. These stresses activate autophagy in order to recycle ATP and maintain survival of the cancerous cells. Autophagy has been shown to enable continued growth of tumor cells by maintaining cellular energy production. By inhibiting autophagy genes in these tumors cells, regression of the tumor and extended survival of the organs affected by the tumors were found. Furthermore, inhibition of autophagy has also been shown to enhance the effectiveness of anticancer therapies.
Mechanism of cell death
Cells that undergo an extreme amount of stress experience cell death either through apoptosis or necrosis. Prolonged autophagy activation leads to a high turnover rate of proteins and organelles. A high rate above the survival threshold may kill cancer cells with a high apoptotic threshold. This technique can be utilized as a therapeutic cancer treatment.
Therapeutic target
New developments in research have found that targeted autophagy may be a viable therapeutic solution in fighting cancer. As discussed above, autophagy plays both a role in tumor suppression and tumor cell survival. Thus, the qualities of autophagy can be used as a strategy for cancer prevention. The first strategy is to induce autophagy and enhance its tumor suppression attributes. The second strategy is to inhibit autophagy and thus induce apoptosis.
The first strategy has been tested by looking at dose-response anti-tumor effects during autophagy-induced therapies. These therapies have shown that autophagy increases in a dose-dependent manner. This is directly related to the growth of cancer cells in a dose-dependent manner as well. This data supports the development of therapies that will encourage autophagy. Secondly, inhibiting the protein pathways directly known to induce autophagy may also serve as an anticancer therapy.
The second strategy is based on the idea that autophagy is a protein degradation system used to maintain homeostasis and the findings that inhibition of autophagy often leads to apoptosis. Inhibition of autophagy is riskier as it may lead to cell survival instead of the desired cell death.
Autophagy and Parkinson disease
Parkinson disease is a neurodegenerative disorder partially caused by the cell death of brain and brain stem cells in many nuclei like the substantia nigra. There are several genetic mutations implicated in the disease, including loss of function PINK1 and Parkin. Loss of function in these genes can lead to damaged mitochondrial accumulation and protein aggregates than can lead to cellular degeneration. Mitochondria is involved in Parkinson’s disease. In idiopathic Parkinson’s disease, the disease is commonly caused by dysfunctional mitochondria, cellular oxidative stress, autophagic alterations and the aggregation of proteins. These can lead to mitochondrial swelling and depolarization.