
 | ||
Tumour heterogeneity describes the observation that different tumour cells can show distinct morphological and phenotypic profiles, including cellular morphology, gene expression, metabolism, motility, proliferation, and metastatic potential. This phenomenon occurs both between tumours (inter-tumour heterogeneity) and within tumours (intra-tumour heterogeneity). A minimal level of intra-tumour heterogeneity is a simple consequence of the imperfection of DNA replication: whenever a cell (normal or cancerous) divides, a few mutations are acquired - leading to a diverse population of cancer cells. The heterogeneity of cancer cells introduces significant challenges in designing effective treatment strategies. However, research into understanding and characterizing heterogeneity can allow for a better understanding of the causes and progression of disease. In turn, this has the potential to guide the creation of more refined treatment strategies that incorporate knowledge of heterogeneity to yield higher efficacy.
Contents
- Models of Heterogeneity
- Cancer Stem Cells
- Clonal Evolution
- Linear expansion
- Branched expansion
- Types Causes of Heterogeneity
- Genetic Heterogeneity
- Other Heterogeneity
- Tumour microenvironment
- Treatment resistance
- Biomarker Discovery
- Model Systems
- Current Strategies
- Experimental
- Sequencing
- References
Tumour heterogeneity has been observed in leukemias, breast, prostate, colon, brain, esophagus, head and neck, bladder and gynecological carcinomas, liposarcoma, and multiple myeloma.
Models of Heterogeneity
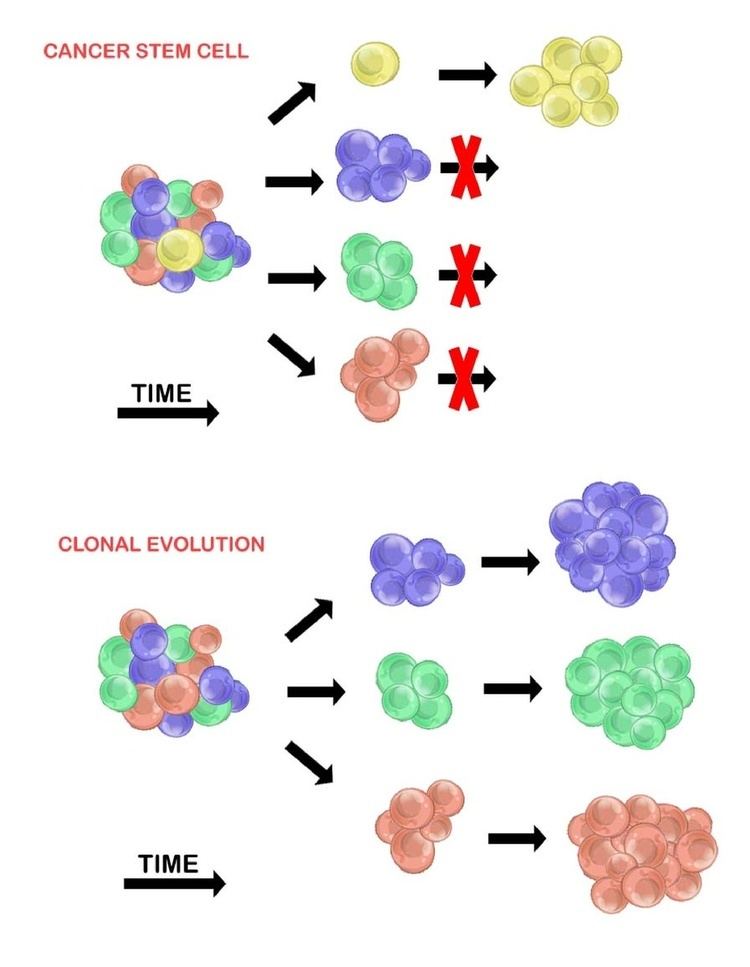
There are two models used to explain the heterogeneity of tumour cells. These are the cancer stem cell model and the clonal evolution model. The models are not mutually exclusive, and it is believed that they both contribute to heterogeneity in varying amounts across different tumour types.
Cancer Stem Cells
The cancer stem cell model asserts that within a population of tumour cells, there is only a small subset of cells that are tumourigenic (able to form tumours). These cells are termed cancer stem cells (CSCs), and are marked by the ability to both self-renew and differentiate into non-tumourigenic progeny. The CSC model posits that the heterogeneity observed between tumour cells is the result of differences in the stem cells from which they originated. Stem cell variability is often caused by epigenetic changes, but can also result from clonal evolution of the CSC population where advantageous genetic mutations can accumulate in CSCs and their progeny (see below).
Evidence of the cancer stem cell model has been demonstrated in multiple tumour types including leukemias, glioblastoma, breast cancer, and prostate cancer.
However, the existence of CSCs is still under debate. One reason for this is that markers for CSCs have been difficult to reproduce across multiple tumours. Further, methods for determining tumourigenic potential utilize xenograft models. These methods suffer from inherent limitations such as the need to control immune response in the transplant animal, and the significant difference in environmental conditions from the primary tumour site to the xenograft site (e.g. absence of required exogenous molecules or cofactors). This has caused some doubt about the accuracy of CSC results and the conclusions about which cells have tumourigenic potential.
Clonal Evolution
The clonal evolution model was first proposed in 1976 by Peter Nowell. In this model, tumours arise from a single mutated cell, accumulating additional mutations as it progresses. These changes give rise to additional subpopulations, and each of these subpopulations has the ability to divide and mutate further. This heterogeneity may give rise to subclones that possess an evolutionary advantage over the others within the tumour environment, and these subclones may become dominant in the tumour over time. When proposed, this model allowed for the understanding of tumour growth, treatment failure, and tumour aggression that occurs during the natural process of tumour formation.
Evolution of the initial tumour cell may occur by two methods:
Linear expansion
Sequentially ordered mutations accumulate in driver genes, tumour suppressor genes, and DNA repair enzymes, resulting in clonal expansion of tumour cells. Linear expansion is less likely to reflect the endpoint of a malignant tumour because the accumulation of mutations is stochastic in heterogeneic tumours.
Branched expansion
Expansion into multiple subclonal populations occurs through a splitting mechanism. This method is more associated with tumour heterogeneity than linear expansion. The acquisition of mutations is random as a result of increased genomic instability with each successive generation. The long-term mutational accumulation may provide a selective advantage during certain stages of tumour progression. The tumour microenvironment may also contribute to tumour expansion, as it is capable of altering the selective pressures that the tumour cells are exposed to.
Types & Causes of Heterogeneity
Multiple types of heterogeneity have been observed between tumour cells, stemming from both genetic and non-genetic variability.
Genetic Heterogeneity
Genetic heterogeneity is a common feature of tumour genomes, and can arise from multiple sources. Some cancers are initiated when exogenous factors introduce mutations, such as ultraviolet radiation (skin cancers) and tobacco (lung cancer). A more common source is genomic instability, which often arises when key regulatory pathways are disrupted in the cells. Some examples include impaired DNA repair mechanisms which can lead to increased replication errors, and defects in the mitosis machinery that allow for large-scale gain or loss of entire chromosomes. Furthermore, it is possible for genetic variability to be further increased by some cancer therapies (e.g. treatment with temozolomide and other chemotherapy drugs).
Other Heterogeneity
Tumour cells can also show heterogeneity between their expression profiles. This is often caused by underlying epigenetic changes. Variation in expression signatures have been detected in different regions of tumour samples within an individual. Researchers have shown that convergent mutations affecting H3K36 methyltransferase SETD2 and histone H3K4 demethylase KDM5C arose in spatially separated tumour sections. Similarly, MTOR, a gene encoding a cell regulatory kinase, has shown to be constitutively active, thereby increasing S6 phosphorylation. This active phosphorylation may serve as a biomarker in clear-cell carcinoma.
Mechanochemical heterogeneity is a hallmark of living eukaryotic cells. It has an impact on epigenetic gene regulation. The heterogeneous dynamic mechanochemical processes regulate interrelationships within the group of cellular surfaces through adhesion. Tumour development and spreading is accompanied by change in heterogeneous chaotic dynamics of mechanochemical interaction process in the group cells, including cells within tumour, and is hierarchical for the host of cancer patients. It is suggested that the heterogeneity of hypoxia in solid tumors is due to the mechanochemical reactions with oxygen nanobubbles. The biological phenomena of mechanochemical heterogeneity maybe used for differential gastric cancer diagnostics against patients with inflammation of gastric mucosa and for increasing antimetastatic activity of dendritic cells based on vaccines when mechanically heterogenized microparticles of tumor cells are used for their loading.There is also a possible methodical approach based on the simultaneous ultrasound imaging diagnostic techniques and therapy, regarding the mechanochemical effect on nanobubles conglomerates with drugs in the tumour.
Tumour microenvironment
Heterogeneity between tumour cells can be further increased due to heterogeneity in the tumour microenvironment. Regional differences in the tumour (e.g. availability of oxygen) impose different selective pressures on tumour cells, leading to a wider spectrum of dominant subclones in different spatial regions of the tumour. The influence of microenvironment on clonal dominance is also a likely reason for the heterogeneity between primary and metastatic tumours seen in many patients, as well as the inter-tumour heterogeneity observed between patients with the same tumour type.
Treatment resistance
Heterogeneic tumours may exhibit different sensitivities to cytotoxic drugs among different clonal populations. This is attributed to clonal interactions that may inhibit or alter therapeutic efficacy, posing a challenge for successful therapies in heterogeneic tumours (and their heterogeneic metastases).
Drug administration in heterogeneic tumours will seldom kill all tumour cells. The initial heterogeneic tumour population may bottleneck, such that few drug resistant cells (if any) will survive. This allows resistant tumour populations to replicate and grow a new tumour through the branching evolution mechanism (see above). The resulting repopulated tumour is heterogeneic and resistant to the initial drug therapy used. The repopulated tumour may also return in a more aggressive manner.
The administration of cytotoxic drugs often results in initial tumour shrinkage. This represents the destruction of initial non-resistant subclonal populations within a heterogeneic tumour, leaving only resistant clones. These resistant clones now contain a selective advantage and can replicate to repopulate the tumour. Replication will likely occur through branching evolution, contributing to tumour heterogeneity. The repopulated tumour may appear to be more aggressive. This is attributed to the drug-resistant selective advantage of the tumour cells.
Biomarker Discovery
Due to the genetic differences within and between tumours, biomarkers that may predict treatment response or prognosis may not be widely applicable. However, it has been suggested that the level of heterogeneity can itself be used as a biomarker since more heterogeneous tumours may be more likely to contain treatment-resistant subclones. Further research into developing biomarkers that account for heterogeneity is still in progress.
Model Systems
Current model systems typically lack the heterogeneity seen in human cancers. In order to accurately study tumour heterogeneity, we must develop more accurate preclinical models. One such model, the patient derived tumour xenograft, has shown excellent utility in preserving tumour heterogeneity whilst allowing detailed study of the drivers of clonal fitness. However, even this model cannot capture the full complexity of cancer.
Current Strategies
While the problem of identifying, characterizing, and treating tumour heterogeneity is still under active research, some effective strategies have been proposed, including both experimental and computational solutions.