
 | ||
What is epigenomics and what it means for our health
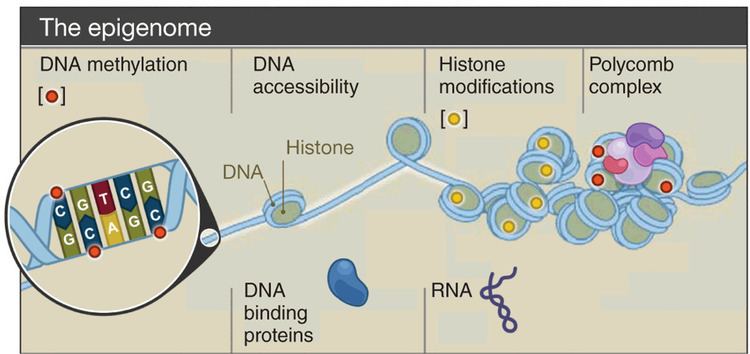
Epigenomics is the study of the complete set of epigenetic modifications on the genetic material of a cell, known as the epigenome. The field is analogous to genomics and proteomics, which are the study of the genome and proteome of a cell. Epigenetic modifications are reversible modifications on a cell’s DNA or histones that affect gene expression without altering the DNA sequence. Epigenomic maintenance is a continuous process and plays an important role in stability of eukaryotic genomes by taking part in crucial biological mechanisms like DNA repair. Plant flavones are said to be inhibiting epigenomic marks that cause cancers. Two of the most characterized epigenetic modifications are DNA methylation and histone modification. Epigenetic modifications play an important role in gene expression and regulation, and are involved in numerous cellular processes such as in differentiation/development and tumorigenesis. The study of epigenetics on a global level has been made possible only recently through the adaptation of genomic high-throughput assays.
Contents
- What is epigenomics and what it means for our health
- video 5 of 8 epigenomics your epigenome and environment
- Introduction to Epigenetics
- Epigenetics
- DNA methylation
- Histone Modification
- Relation to other genomic fields
- Histone modification assays
- ChIP Chip and ChIP Seq
- DNA Methylation assays
- Restriction endonuclease based methods
- Bisulfite sequencing
- Direct Detection
- Theoretical modeling approaches
- References
video 5 of 8 epigenomics your epigenome and environment
Introduction to Epigenetics
The mechanisms governing phenotypic plasticity, or the capacity of a cell to change its state in response to stimuli, have long been the subject of research (Phenotypic plasticity 1). The traditional central dogma of biology states that the DNA of a cell is transcribed to RNA, which is translated to proteins, which perform cellular processes and functions. A paradox exists, however, in that cells exhibit diverse responses to varying stimuli and that cells sharing identical sets of DNA such as in multicellular organisms can have a variety of distinct functions and phenotypes. Classical views have attributed phenotypic variation to differences in primary DNA structure, be it through aberrant mutation or an inherited sequence allele. However, while this did explain some aspects of variation, it does not explain how tightly coordinated and regulated cellular responses, such as differentiation, are carried out.
A more likely source of cellular plasticity is through the Regulation of gene expression, such that while two cells may have near identical DNA, the differential expression of certain genes results in variation. Research has shown that cells are capable of regulating gene expression at several stages: mRNA transcription, processing and transportation as well as in protein translation, post-translational processing and degradation. Regulatory proteins that bind to DNA, RNA, and/or proteins are key effectors in these processes and function by positively or negatively regulating specific protein level and function in a cell. And, while DNA binding transcription factors provide a mechanism for specific control of cellular responses, a model where DNA binding transcription factors are the sole regulators of gene activity is also unlikely. For example, in a study of Somatic-cell nuclear transfer, it was demonstrated that stable features of differentiation remain after the nucleus is transferred to a new cellular environment, suggesting that a stable and heritable mechanism of gene regulation was involved in the maintenance of the differentiated state in the absence of the DNA binding transcription factors.
With the finding that DNA methylation and histone modifications are stable, heritable, and also reversible processes that influence gene expression without altering DNA primary structure, a mechanism for the observed variability in cell gene expression was provided. These modifications were termed epigenetic, from epi “on top of” the genetic material “DNA” (Epigenetics 1). The mechanisms governing epigenetic modifications are complex, but through the advent of high-throughput sequencing technology they are now becoming better understood.
Epigenetics
Genomic modifications that alter gene expression that cannot be attributed to modification of the primary DNA sequence and that are heritable mitotically and meiotically are classified as epigenetic modifications. DNA methylation and histone modification are among the best characterized epigenetic processes.
DNA methylation
The first epigenetic modification to be characterized in depth was DNA methylation. As its name implies, DNA methylation is the process by which a methyl group is added to DNA. The enzymes responsible for catalyzing this reaction are the DNA methyltransferases (DNMTs). While DNA methylation is stable and heritable, it can be reversed by an antagonistic group of enzymes known as DNA de-methylases. In eukaryotes, methylation is most commonly found on the carbon 5 position of cytosine residues (5mC) adjacent to guanine, termed CpG dinucleotides.
DNA methylation patterns vary greatly between species and even within the same organism. The usage of methylation among animals is quite different; with vertebrates exhibiting the highest levels of 5mC and invertebrates more moderate levels of 5mC. Some organisms like Caenorhabditis elegans have not been demonstrated to have 5mC nor a conventional DNA methyltransferase; this would suggest that other mechanisms other than DNA methylation are also involved.
Within an organism, DNA methylation levels can also vary throughout development and by region. For example, in mouse primordial germ cells, a genome wide de-methylation even occurs; by implantation stage, methylation levels return to their prior somatic levels. When DNA methylation occurs at promoter regions, the sites of transcription initiation, it has the effect of repressing gene expression. This is in contrast to unmethylated promoter regions which are associated with actively expressed genes.
The mechanism by which DNA methylation represses gene expression is a multi-step process. The distinction between methylated and unmethylated cytosine residues is carried out by specific DNA-binding proteins. Binding of these proteins recruit histone deacetylases (HDACs) enzyme which initiate chromatin remodeling such that the DNA becoming less accessible to transcriptional machinery, such as RNA polymerase, effectively repressing gene expression.
Histone Modification
In eukaryotes, genomic DNA is coiled into protein-DNA complexes called chromatin. Histones, which are the most prevalent type of protein found in chromatin, function to condense the DNA; the net positive charge on histones facilitates their bonding with DNA, which is negatively charged. The basic and repeating units of chromatin, nucleosomes, consist of an octamer of histone proteins (H2A, H2B, H3 and H4) and a 146 bp length of DNA wrapped around it. Nucleosomes and the DNA connecting form a 10 nm diameter chromatin fiber, which can be further condensed.
Chromatin packaging of DNA varies depending on the cell cycle stage and by local DNA region. The degree to which chromatin is condensed is associated with a certain transcriptional state. Unpackaged or loose chromatin is more transcriptionally active than tightly packaged chromatin because it is more accessible to transcriptional machinery. By remodeling chromatin structure and changing the density of DNA packaging, gene expression can thus be modulated.
Chromatin remodeling occurs via post-translational modifications of the N-terminal tails of core histone proteins. The collective set of histone modifications in a given cell is known as the histone code. Many different types of histone modification are known, including: acetylation, methylation, phosphorylation, ubiquitination, SUMOylation, ADP-ribosylation, deamination and proline isomerization; acetylation, methylation, phosphorylation and ubiquitination have been implicated in gene activation whereas methylation, ubiquitination, SUMOylation, deamination and proline isomerization have been implicated in gene repression. Note that several modification types including methylation, phosphorylation and ubiquitination can be associated with different transcriptional states depending on the specific amino acid on the histone being modified. Furthermore, the DNA region where histone modification occurs can also elicit different effects; an example being methylation of the 3rd core histone at lysine residue 36 (H3K36). When H3K36 occurs in the coding sections of a gene, it is associated with gene activation but the opposite is found when it is within the promoter region.
Histone modifications regulate gene expression by two mechanisms: by disruption of the contact between nucleosomes and by recruiting chromatin remodeling ATPases. An example of the first mechanism occurs during the acetylation of lysine terminal tail amino acids, which is catalyzed by histone acetyltransferases (HATs). HATs are part of a multiprotein complex that is recruited to chromatin when activators bind to DNA binding sites. Acetylation effectively neutralizes the basic charge on lysine, which was involved in stabilizing chromatin through its affinity for negatively charged DNA. Acetylated histones therefore favor the dissociation of nucleosomes and thus unwinding of chromatin can occur. Under a loose chromatin state, DNA is more accessible to transcriptional machinery and thus expression is activated. The process can be reversed through removal of histone acetyl groups by deacetylases.
The second process involves the recruitment of chromatin remodeling complexes by the binding of activator molecules to corresponding enhancer regions. The nucleosome remodeling complexes reposition nucleosomes by several mechanisms, enabling or disabling accessibility of transcriptional machinery to DNA. The SWI/SNF protein complex in yeast is one example of a chromatin remodeling complex that regulates the expression of many genes through chromatin remodeling.
Relation to other genomic fields
Epigenomics shares many commonalities with other genomics fields, in both methodology and in its abstract purpose. Epigenomics seeks to identify and characterize epigenetic modifications on a global level, similar to the study of the complete set of DNA in genomics or the complete set of proteins in a cell in proteomics. The logic behind performing epigenetic analysis on a global level is that inferences can be made about epigenetic modifications, which might not otherwise be possible through analysis of specific loci. As in the other genomics fields, epigenomics relies heavily on bioinformatics, which combines the disciplines of biology, mathematics and computer science. However while epigenetic modifications had been known and studied for decades, it is through these advancements in bioinformatics technology that have allowed analyses on a global scale. Many current techniques still draw on older methods, often adapting them to genomic assays as is described in the next section.
Histone modification assays
The cellular processes of transcription, DNA replication and DNA repair involve the interaction between genomic DNA and nuclear proteins. It had been known that certain regions within chromatin were extremely susceptible to DNAse I digestion, which cleaves DNA in a low sequence specificity manner. Such hypersensitive sites were thought to be transcriptionally active regions, as evidenced by their association with RNA polymerase and topoisomerases I and II.
It is now known that sensitivity to DNAse I regions correspond to regions of chromatin with loose DNA-histone association. Hypersensitive sites most often represent promoters regions, which require for DNA to be accessible for DNA binding transcriptional machinery to function.
ChIP-Chip and ChIP-Seq
Histone modification was first detected on a genome wide level through the coupling of chromatin immunoprecipitation (ChIP) technology with DNA microarrays, termed ChIP-Chip. However instead of isolating a DNA-binding transcription factor or enhancer protein through chromatin immunoprecipitation, the proteins of interest are the modified histones themselves. First, histones are cross-linked to DNA in vivo through light chemical treatment (e.g., formaldehyde). The cells are next lysed, allowing for the chromatin to be extracted and fragmented, either by sonication or treatment with a non-specific restriction enzyme (e.g., micrococcal nuclease). Modification-specific antibodies in turn, are used to immunoprecipitate the DNA-histone complexes. Following immunoprecipitation, the DNA is purified from the histones, amplified via PCR and labeled with a fluorescent tag (e.g., Cy5, Cy3). The final step involves hybridization of labeled DNA, both immunoprecipitated DNA and non-immunoprecipitated onto a microarray containing immobilized gDNA. Analysis of the relative signal intensity allows the sites of histone modification to be determined.
ChIP-chip was used extensively to characterize the global histone modification patterns of yeast. From these studies, inferences on the function of histone modifications were made; that transcriptional activation or repression was associated with certain histone modifications and by region. While this method was effective providing near full coverage of the yeast epigenome, its use in larger genomes such as humans is limited.
In order to study histone modifications on a truly genome level, other high-throughput methods were coupled with the chromatin immunoprecipitation, namely: SAGE: serial analysis of gene expression (ChIP-SAGE), PET: paired end ditag sequencing (ChIP-PET) and more recently, next-generation sequencing (ChIP-Seq). ChIP-seq follows the same protocol for chromatin immunoprecipitation but instead of amplification of purified DNA and hybridization to a microarray, the DNA fragments are directly sequenced using next generation parallel re-sequencing. It has proven to be an effective method for analyzing the global histone modification patterns and protein target sites, providing higher resolution than previous methods.
DNA Methylation assays
Techniques for characterizing primary DNA sequences could not be directly applied to methylation assays. For example, when DNA was amplified in PCR or bacterial cloning techniques, the methylation pattern was not copied and thus the information lost. The DNA hybridization technique used in DNA assays, in which radioactive probes were used to map and identify DNA sequences, could not be used to distinguish between methylated and non-methylated DNA.
Restriction endonuclease based methods
Non genome-wide approaches
The earliest methylation detection assays used methylation modification sensitive restriction endonucleases. Genomic DNA was digested with both methylation-sensitive and insensitive restriction enzymes recognizing the same restriction site. The idea being that whenever the site was methylated, only the methylation insensitive enzyme could cleave at that position. By comparing restriction fragment sizes generated from the methylation-sensitive enzyme to those of the methylation-insensitive enzyme, it was possible to determine the methylation pattern of the region. This analysis step was done by amplifying the restriction fragments via PCR, separating them through gel electrophoresis and analyzing them via southern blot with probes for the region of interest.
This technique was used to compare the DNA methylation modification patterns in the human adult and hemoglobin gene loci. Different regions of the gene (gamma delta beta globin) were known to be expressed at different stages of development. Consistent with a role of DNA methylation in gene repression, regions that were associated with high levels of DNA methylation were not actively expressed.
This method was limited not suitable for studies on the global methylation pattern, or ‘methylome’. Even within specific loci it was not fully representative of the true methylation pattern as only those restriction sites with corresponding methylation sensitive and insensitive restriction assays could provide useful information. Further complications could arise when incomplete digestion of DNA by restriction enzymes generated false negative results.
Genome wide approaches
DNA methylation profiling on a large scale was first made possible through the Restriction Landmark Genome Scanning (RLGS) technique. Like the locus-specific DNA methylation assay, the technique identified methylated DNA via its digestion methylation sensitive enzymes. However it was the use of two-dimensional gel electrophoresis that allowed be characterized on a broader scale.
However it was not until the advent of microarray and next generation sequencing technology when truly high resolution and genome-wide DNA methylation became possible. As with RLGS, the endonuclease component is retained in the method but it is coupled to new technologies. One such approach is the differential methylation hybridization (DMH), in which one set of genomic DNA is digested with methylation-sensitive restriction enzymes and a parallel set of DNA is not digested. Both sets of DNA are subsequently amplified and each labelled with fluorescent dyes and used in two-colour array hybridization. The level of DNA methylation at a given loci is determined by the relative intensity ratios of the two dyes. Adaptation of next generation sequencing to DNA methylation assay provides several advantages over array hybridization. Sequence-based technology provides higher resolution to allele specific DNA methylation, can be performed on larger genomes, and does not require creation of DNA microarrays which require adjustments based on CpG density to properly function.
Bisulfite sequencing
Bisulfite sequencing relies on chemical conversion of unmethylated cytosines exclusively, such that they can be identified through standard DNA sequencing techniques. Sodium bisulfate and alkaline treatment does this by converting unmethylated cytosine residues into uracil while leaving methylated cytosine unaltered. Subsequent amplification and sequencing of untreated DNA and sodium bisulphite treated DNA allows for methylated sites to be identified. Bisulfite sequencing, like the traditional restriction based methods, was historically limited to methylation patterns of specific gene loci, until whole genome sequencing technologies became available. However, unlike traditional restriction based methods, bisulfite sequencing provided resolution on a nucleotide level.
Limitations of the bisulfite technique include the incomplete conversion of cytosine to uracil, which is a source of false positives. Further, bisulfite treatment also causes DNA degradation and requires an additional purification step to remove the sodium bisulfite.
Next-generation sequencing is well suited in complementing bisulfite sequencing in genome-wide methylation analysis. While this now allows for methylation pattern to be determined on the highest resolution possible, on the single nucleotide level, challenges still remain in the assembly step because of reduced sequence complexity in bisulphite treated DNA. Increases in read length seek to address this challenge, allowing for whole genome shotgun bisulphite sequencing (WGBS) to be performed. The WGBS approach using an Illumina Genome Analyzer platform and has already been implemented in Arabidopsis thaliana.
Direct Detection
Polymerase sensitivity in single molecule real time sequencing made it possible for scientists to directly detect epigenetic marks such as methylation as the polymerase moves along the DNA molecule being sequenced. Several projects have demonstrated the ability to collect genome-wide epigenetic data in bacteria.
Theoretical modeling approaches
First mathematical models for different nucleosome states affecting gene expression were introduced in 1980s [ref]. Later, this idea was almost forgotten, until the experimental evidence has indicated a possible role of covalent histone modifications as an epigenetic code. In the next several years, high-throughput data have indeed uncovered the abundance of epigenetic modifications and their relation to chromatin functioning which has motivated new theoretical models for the appearance, maintaining and changing these patterns,. These models are usually formulated in the frame of one-dimensional lattice approaches.