
 | ||
Mathematical and theoretical biology is an interdisciplinary scientific research field with a range of applications. The field is sometimes called mathematical biology or biomathematics to stress the mathematical side, or theoretical biology to stress the biological side. Theoretical biology focuses more on the development of theoretical principles for biology while mathematical biology focuses on the use of mathematical tools to study biological systems, even though the two terms are sometimes interchanged. Mathematical biology aims at the mathematical representation, treatment and modeling of biological processes, using techniques and tools of applied mathematics. It has both theoretical and practical applications in biological, biomedical and biotechnology research. Describing systems in a quantitative manner means their behavior can be better simulated, and hence properties can be predicted that might not be evident to the experimenter. This requires precise mathematical models.
Contents
- Importance
- Areas of research
- Evolutionary biology
- Computer models and automata theory
- Molecular set theory
- Mathematical methods
- Simulation of mathematical biology
- Mathematical biophysics
- Deterministic processes dynamical systems
- Stochastic processes random dynamical systems
- Spatial modelling
- Organizational biology
- Algebraic biology
- Model example the cell cycle
- Important names
- References
Mathematical biology employs many components of mathematics, and has contributed to the development of new techniques.
Importance
Applying mathematics to biology has a long history, but only recently has there been an explosion of interest in the field. Some reasons for this include:
Areas of research
Several areas of specialized research in mathematical and theoretical biology as well as external links to related projects in various universities are concisely presented in the following subsections, including also a large number of appropriate validating references from a list of several thousands of published authors contributing to this field. Many of the included examples are characterised by highly complex, nonlinear, and supercomplex mechanisms, as it is being increasingly recognised that the result of such interactions may only be understood through a combination of mathematical, logical, physical/chemical, molecular and computational models. Due to the wide diversity of specific knowledge involved, biomathematical research is often done in collaboration between mathematicians, biomathematicians, theoretical biologists, bioinformaticians, biostatisticians, physicists, biophysicists, biochemists, bioengineers, engineers, biologists, physiologists, research physicians, biomedical researchers, oncologists, molecular biologists, geneticists, embryologists, zoologists, chemists, etc.
Evolutionary biology
Ecology and evolutionary biology have traditionally been the dominant fields of mathematical biology.
Evolutionary biology has been the subject of extensive mathematical theorizing. The traditional approach in this area, which includes complications from genetics, is population genetics. Most population geneticists consider the appearance of new alleles by mutation, the appearance of new genotypes by recombination, and changes in the frequencies of existing alleles and genotypes at a small number of gene loci. When infinitesimal effects at a large number of gene loci are considered, together with the assumption of linkage equilibrium or quasi-linkage equilibrium, one derives quantitative genetics. Ronald Fisher made fundamental advances in statistics, such as analysis of variance, via his work on quantitative genetics. Another important branch of population genetics that led to the extensive development of coalescent theory is phylogenetics. Phylogenetics is an area that deals with the reconstruction and analysis of phylogenetic (evolutionary) trees and networks based on inherited characteristics Traditional population genetic models deal with alleles and genotypes, and are frequently stochastic.
Many population genetics models assume that population sizes are constant. Variable population sizes, often in the absence of genetic variation, are treated by the field of population dynamics. Work in this area dates back to the 19th century, and even as far as 1798 when Thomas Malthus formulated the first principle of population dynamics, which later became known as the Malthusian growth model. The Lotka–Volterra predator-prey equations are another famous example. Population dynamics overlap with another active area of research in mathematical biology: mathematical epidemiology, the study of infectious disease affecting populations. Various models of the spread of infections have been proposed and analyzed, and provide important results that may be applied to health policy decisions.
In evolutionary game theory, developed first by John Maynard Smith and George R. Price, selection acts directly on inherited phenotypes, without genetic complications. This approach has been mathematically refined to produce the field of adaptive dynamics.
Computer models and automata theory
A monograph on this topic summarizes an extensive amount of published research in this area up to 1986, including subsections in the following areas: computer modeling in biology and medicine, arterial system models, neuron models, biochemical and oscillation networks, quantum automata, quantum computers in molecular biology and genetics, cancer modelling, neural nets, genetic networks, abstract categories in relational biology, metabolic-replication systems, category theory applications in biology and medicine, automata theory, cellular automata, tessellation models and complete self-reproduction, chaotic systems in organisms, relational biology and organismic theories. This published report also includes 390 references to peer-reviewed articles by a large number of authors.
Modeling cell and molecular biology
This area has received a boost due to the growing importance of molecular biology.
Modelling physiological systems
Molecular set theory
Molecular set theory (MST) is a mathematical formulation of the wide-sense chemical kinetics of biomolecular reactions in terms of sets of molecules and their chemical transformations represented by set-theoretical mappings between molecular sets. It was introduced by Anthony Bartholomay, and its applications were developed in mathematical biology and especially in mathematical medicine. In a more general sense, MST is the theory of molecular categories defined as categories of molecular sets and their chemical transformations represented as set-theoretical mappings of molecular sets. The theory has also contributed to biostatistics and the formulation of clinical biochemistry problems in mathematical formulations of pathological, biochemical changes of interest to Physiology, Clinical Biochemistry and Medicine.
Mathematical methods
A model of a biological system is converted into a system of equations, although the word 'model' is often used synonymously with the system of corresponding equations. The solution of the equations, by either analytical or numerical means, describes how the biological system behaves either over time or at equilibrium. There are many different types of equations and the type of behavior that can occur is dependent on both the model and the equations used. The model often makes assumptions about the system. The equations may also make assumptions about the nature of what may occur.
Simulation of mathematical biology
Computer with significant recent evolution in performance acceraretes the model simulation based on various formulas. The websites BioMath Modeler can run simulations and display charts interactively on browser.
Mathematical biophysics
The earlier stages of mathematical biology were dominated by mathematical biophysics, described as the application of mathematics in biophysics, often involving specific physical/mathematical models of biosystems and their components or compartments.
The following is a list of mathematical descriptions and their assumptions.
Deterministic processes (dynamical systems)
A fixed mapping between an initial state and a final state. Starting from an initial condition and moving forward in time, a deterministic process always generates the same trajectory, and no two trajectories cross in state space.
Stochastic processes (random dynamical systems)
A random mapping between an initial state and a final state, making the state of the system a random variable with a corresponding probability distribution.
Spatial modelling
One classic work in this area is Alan Turing's paper on morphogenesis entitled The Chemical Basis of Morphogenesis, published in 1952 in the Philosophical Transactions of the Royal Society.
Organizational biology
Theoretical approaches to biological organization aim to understand the interdependence between the parts of organisms. They emphasize the circularities that these interdependences lead to. Theoretical biologists developed several concepts to formalize this idea.
For example, abstract relational biology (ARB) is concerned with the study of general, relational models of complex biological systems, usually abstracting out specific morphological, or anatomical, structures. Some of the simplest models in ARB are the Metabolic-Replication, or (M,R)--systems introduced by Robert Rosen in 1957-1958 as abstract, relational models of cellular and organismal organization.
Other approaches include th notion of autopoiesis developed by Maturana and Varela, Kauffman's Work-Constraints cycles, and more recently the notion of closure of constraints.
Algebraic biology
Algebraic biology (also known as symbolic systems biology) applies the algebraic methods of symbolic computation to the study of biological problems, especially in genomics, proteomics, analysis of molecular structures and study of genes.
Model example: the cell cycle
The eukaryotic cell cycle is very complex and is one of the most studied topics, since its misregulation leads to cancers. It is possibly a good example of a mathematical model as it deals with simple calculus but gives valid results. Two research groups have produced several models of the cell cycle simulating several organisms. They have recently produced a generic eukaryotic cell cycle model that can represent a particular eukaryote depending on the values of the parameters, demonstrating that the idiosyncrasies of the individual cell cycles are due to different protein concentrations and affinities, while the underlying mechanisms are conserved (Csikasz-Nagy et al., 2006).
By means of a system of ordinary differential equations these models show the change in time (dynamical system) of the protein inside a single typical cell; this type of model is called a deterministic process (whereas a model describing a statistical distribution of protein concentrations in a population of cells is called a stochastic process).
To obtain these equations an iterative series of steps must be done: first the several models and observations are combined to form a consensus diagram and the appropriate kinetic laws are chosen to write the differential equations, such as rate kinetics for stoichiometric reactions, Michaelis-Menten kinetics for enzyme substrate reactions and Goldbeter–Koshland kinetics for ultrasensitive transcription factors, afterwards the parameters of the equations (rate constants, enzyme efficiency coefficients and Michaelis constants) must be fitted to match observations; when they cannot be fitted the kinetic equation is revised and when that is not possible the wiring diagram is modified. The parameters are fitted and validated using observations of both wild type and mutants, such as protein half-life and cell size.
To fit the parameters, the differential equations must be studied. This can be done either by simulation or by analysis. In a simulation, given a starting vector (list of the values of the variables), the progression of the system is calculated by solving the equations at each time-frame in small increments.
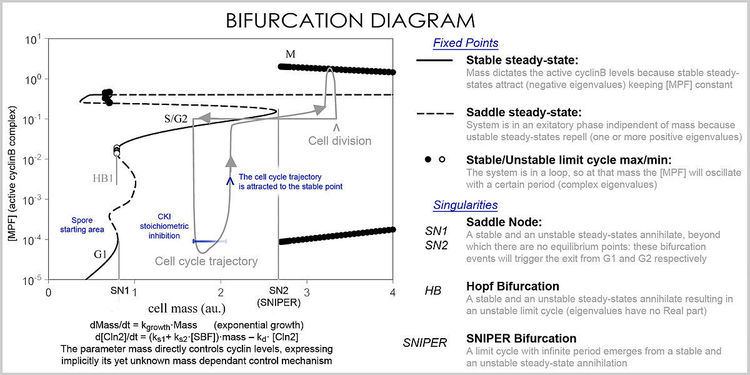
In analysis, the properties of the equations are used to investigate the behavior of the system depending of the values of the parameters and variables. A system of differential equations can be represented as a vector field, where each vector described the change (in concentration of two or more protein) determining where and how fast the trajectory (simulation) is heading. Vector fields can have several special points: a stable point, called a sink, that attracts in all directions (forcing the concentrations to be at a certain value), an unstable point, either a source or a saddle point, which repels (forcing the concentrations to change away from a certain value), and a limit cycle, a closed trajectory towards which several trajectories spiral towards (making the concentrations oscillate).
A better representation, which handles the large number of variables and parameters, is a bifurcation diagram using bifurcation theory. The presence of these special steady-state points at certain values of a parameter (e.g. mass) is represented by a point and once the parameter passes a certain value, a qualitative change occurs, called a bifurcation, in which the nature of the space changes, with profound consequences for the protein concentrations: the cell cycle has phases (partially corresponding to G1 and G2) in which mass, via a stable point, controls cyclin levels, and phases (S and M phases) in which the concentrations change independently, but once the phase has changed at a bifurcation event (Cell cycle checkpoint), the system cannot go back to the previous levels since at the current mass the vector field is profoundly different and the mass cannot be reversed back through the bifurcation event, making a checkpoint irreversible. In particular the S and M checkpoints are regulated by means of special bifurcations called a Hopf bifurcation and an infinite period bifurcation.