
ICD-9-CM 425.1 DiseasesDB 6373 | ICD-10 I42.1–I42.2 OMIM 192600 | |
 | ||
Synonyms Asymmetric septal hypertrophy, idiopathic hypertrophic subaortic stenosis | ||
Hypertrophic cardiomyopathy (HCM) is a disease in which a portion of the myocardium (heart muscle) is hypertrophic (enlarged) without any obvious cause, creating functional impairment of the heart. It is the leading cause of sudden cardiac death in young athletes. The occurrence of hypertrophic cardiomyopathy is a significant cause of sudden cardiac death in any age group and a cause of disabling cardiac symptoms. HCM is frequently asymptomatic until sudden cardiac death, and for this reason some suggest routinely screening certain populations for this disease. In most patients, HCM is associated with little or no disability and normal life expectancy. Diagnosis is usually by echocardiogram. While there is no known prevention or cure, symptomatic patients may be treated effectively by medication or, in severe cases, by surgery.
Contents
- Signs and symptoms
- Genetics
- Pathophysiology
- Dynamic outflow obstruction
- Screening
- United States
- Canada
- UK
- Diagnosis
- Obstructive or non obstructive
- Physical examination
- Cardiac catheterization
- Asymptomatic patients
- Medications
- Surgical septal myectomy
- Alcohol septal ablation
- Mitral clip
- Implantable pacemaker or defibrillator
- Cardiac transplantation
- Prognosis
- Children
- Other animals
- References
A cardiomyopathy is a disease that affects the muscle of the heart. With HCM, the myocytes (cardiac contractile cells) in the heart increase in size, which results in the thickening of the heart muscle. In addition, the normal alignment of muscle cells is disrupted, a phenomenon which is known as myocardial disarray. HCM also causes disruptions of the electrical functions of the heart. British pathologist Donald Teare published the first modern description of the disease in 1958.
HCM is most commonly due to a mutation in one of nine genes that results in a mutated protein in the sarcomere, the primary component of the myocyte (the muscle cell of the heart). These are predominantly single-point missense mutations in the genes for the beta-myosin heavy chain (MHC), myosin-binding protein C, cardiac troponin T, troponin I, or tropomyosin. These mutations cause myofibril and myocyte structural abnormalities and possible deficiencies in force generation. Each child of an HCM parent has 50% chance of inheriting the disease-causing mutation. Family-specific genetic testing can often identify at-risk relatives, although disease severity and age of onset cannot be predicted.
While most literature so far focuses on European, American, and Japanese populations, HCM appears in all ethnic groups. The prevalence of HCM is about 0.2% of the general population, or one in 500. In addition, it is the most common heart disease in domestic cats.
Signs and symptoms
The clinical course of HCM is variable. Many patients are asymptomatic or mildly symptomatic. The symptoms and signs of HCM include shortness of breath (dyspnea) due to stiffening and decreased blood filling of the ventricles, exertional chest pain (sometimes known as angina) due to reduced or restricted blood flow (ischemia) to the coronary arteries, uncomfortable awareness of the heart beat (palpitations) due to the aforementioned ischemia, as well as disruption of the electrical system running through the abnormal heart muscle, lightheadedness, fatigue, fainting (called syncope) and sudden cardiac death. As mentioned, dyspnea is largely due to increased stiffness of the left ventricle (LV), which impairs filling of the ventricles, but also leads to elevated pressure in the left ventricle and left atrium, causing back pressure and interstitial congestion in the lungs. Symptoms are not closely related to the presence or severity of an outflow tract gradient. Often, symptoms mimic those of congestive heart failure (esp. activity intolerance and dyspnea), but treatment of each is different. Beta blockers are used in both cases, but treatment with diuretics, a mainstay of CHF treatment, will exacerbate symptoms in hypertrophic obstructive cardiomyopathy by decreasing ventricular preload volume and thereby increasing outflow resistance (less blood to push aside the thickened obstructing tissue).
Major risk factors for sudden death in individuals with HCM include prior history of cardiac arrest or ventricular fibrillation, spontaneous sustained ventricular tachycardia, family history of premature sudden death, unexplained syncope, LV thickness greater than or equal to 30 mm, abnormal exercise blood pressure and nonsustained ventricular tachycardia.
Genetics
Familial hypertrophic cardiomyopathy is inherited as an autosomal dominant trait and is attributed to mutations in one of a number of genes that encode for the sarcomere proteins.
Currently, about 50–60% of patients with a high index of clinical suspicion for HCM will have a mutation identified in at least one of nine sarcomeric genes. Approximately 40% of these mutations occur in the β-myosin heavy chain gene on chromosome 14 q11.2-3, and approximately 40% involve the cardiac myosin-binding protein C gene. Since HCM is typically an autosomal dominant trait, children of a single HCM parent have 50% chance of inheriting the disease-causing mutation. Whenever such a mutation is identified, family-specific genetic testing can be used to identify relatives at-risk for the disease, although clinical severity and age of onset cannot be predicted.
In individuals without a family history of HCM, the most common cause of the disease is a de novo mutation of the gene that produces the β-myosin heavy chain.
An insertion/deletion polymorphism in the gene encoding for angiotensin converting enzyme (ACE) alters the clinical phenotype of the disease. The D/D (deletion/deletion) genotype of ACE is associated with more marked hypertrophy of the left ventricle and may be associated with higher risk of adverse outcomes.
Some mutations could have more harmful potential compared to others (β-myosin heavy chain). For example, troponin T mutations were originally associated with a 50% mortality before the age of 40. However, a more recent and larger study found a similar risk to other sarcomeric protein mutations.
Pathophysiology
Individuals with HCM have some degree of left ventricular hypertrophy. Usually this is an asymmetric hypertrophy, involving the inter-ventricular septum, and is known as asymmetric septal hypertrophy. This is in contrast to the concentric hypertrophy seen in aortic stenosis or hypertension. About two-thirds of individuals with HCM have asymmetric septal hypertrophy.
About 25% of individuals with HCM demonstrate an obstruction to the outflow of blood from the left ventricle during rest. In about 70% of patients, however, the obstruction can be provoked under certain conditions. This is known as dynamic outflow obstruction because the degree of obstruction is variable and is dependent on the loading conditions (ventricular filling and arterial blood pressure) and the contractility state of the left ventricle.
Myocardial hypertrophy and extracellular fibrosis predispose to increased left ventricular stiffness, which in concert with compromised cellular energetics and abnormal calcium handling, leads to diastolic dysfunction manifested as dyspnea and exercise intolerance.
The altered structure of the coronary vessels and increased diastolic pressure (reduced blood supply), together with the hypertrophy and the outflow tract obstruction (increased demand), cause myocardial ischemia that is manifested as angina and may be responsible for the triggering of ventricular arrhythmias.
In about 30% of patients, there are abnormal vascular responses and inability to increase systolic blood pressure during exercise. This is attributed to exaggerated cardiac inhibitory reflexes initiated by increased myocardial wall stress and to elevated levels of vasodilating substances (natriuretic peptides).
Dynamic outflow obstruction
Dynamic outflow obstruction (when present in HCM) is usually due to systolic anterior motion (SAM) of the anterior leaflet of the mitral valve. Echocardiographic evidence indicates that drag, the pushing force of flow, is the dominant hydrodynamic force on the mitral leaflets. In obstructive HCM, the mitral leaflets are often large and are anteriorly positioned in the LV cavity due to anteriorly positioned papillary muscles that at surgery are often "agglutinated" onto the LV anterior wall by abnormal attachments.
The mid-septal bulge aggravates the malposition of the valve and redirects outflow so that it comes from a lateral and posterior direction. The abnormally directed outflow may be visualized behind and lateral to the enlarged mitral valve, where it catches it and pushes it into the septum. There is a crucial overlap between the inflow and outflow portions of the left ventricle. As SAM progresses in early systole, the angle between outflow and the protruding mitral leaflet increases. A greater surface area of the leaflets is now exposed to drag, which amplifies the force on the leaflets – drag increases with increasing angle relative to flow. An analogy is an open door in a drafty corridor: the door starts by moving slowly and then accelerates as it presents a greater surface area to the wind and finally it slams shut. The necessary conditions that predispose to SAM are: anterior position of the mitral valve in the LV, altered LV geometry that allows flow to strike the mitral valve from behind, and chordal slack. SAM may be considered anteriorly directed mitral prolapse. In both conditions, the mitral valve is enlarged and is displaced in systole by the pushing force of flow resulting in mitral regurgitation.
Because the mitral valve leaflet doesn't get pushed into the left ventricular outflow tract (LVOT) until after the aortic valve opens, the initial upstroke of the arterial pulse will be normal. When the mitral valve leaflet gets pushed into the LVOT, the arterial pulse will momentarily collapse and be followed by a second rise, as the left ventricular pressure overcomes the increased obstruction that SAM of the mitral valve causes. This can be seen on the physical examination as a double tap upon palpation of the apical impulse and as a double pulsation upon palpation of the carotid pulse, known as bifid pulse.
Screening
Although HCM may be asymptomatic, affected individuals may present with symptoms ranging from mild to critical heart failure and sudden cardiac death at any point from early childhood to seniority. HCM is the leading cause of sudden cardiac death in young athletes in the United States, and the most common genetic cardiovascular disorder. One study found that the incidence of sudden cardiac death in young competitive athletes declined in the Veneto region of Italy by 89% since the 1982 introduction of routine cardiac screening for athletes, from an unusually high starting rate. As of 2010, however, studies have shown that the incidence of sudden cardiac death, among all HCM patients, has declined to one percent or less. Screen-positive individuals who are diagnosed with cardiac disease are usually told to avoid competitive athletics.
HCM can be detected with an echocardiogram (ECHO) with 80%+ accuracy, which can be preceded by screening with an electrocardiogram (ECG) to test for heart abnormalities. Cardiac magnetic resonance imaging (CMR), considered the gold standard for determining the physical properties of the left ventricular wall, can serve as an alternative screening tool when an echocardiogram provides inconclusive results. For example, the identification of segmental lateral ventricular hypertrophy cannot be accomplished with echocardiography alone. Also, left ventricular hypertrophy may be absent in children under thirteen years of age. This undermines the results of pre-adolescents’ echocardiograms. Researchers, however, have studied asymptomatic carriers of an HCM-causing mutation through the use of CMR and have been able to identify crypts in the interventricular septal tissue in these patients. It has been proposed that the formation of these crypts is an indication of myocyte disarray and altered vessel walls that may later result in the clinical expression of HCM. Lastly, giving warning of heart abnormalities in only 3% of patients before sudden cardiac death, the gathering of family history and physical examination alone are ineffective. A possible explanation for this is that the typical gathering of family history only focuses on whether sudden death occurred or not. It fails to acknowledge the age at which relatives suffered sudden cardiac death, as well as the frequency of the cardiac events. Furthermore, given the several factors necessary to be considered at risk for sudden cardiac death, while most of the factors do not have strong predictive value individually, there exists ambiguity regarding when to implement special treatment.
United States
There are several potential challenges associated with routine screening for HCM in the United States. First, the U.S. athlete population of 15 million is almost twice as large as Italy's estimated athlete population. Second, these events are rare, with fewer than 100 deaths in the U.S. due to HCM in competitive athletes per year, or about 1 death per 220,000 athletes. Lastly, genetic testing would provide a definitive diagnosis; however, due to the numerous HCM-causing mutations, this method of screening is complex and is not cost-effective. Therefore, genetic testing in the United States is limited to individuals who exhibit clear symptoms of HCM, and their family members. This ensures that the test is not wasted on detecting other causes of ventricular hypertrophy (due to its low sensitivity), and that family members of the individual are educated on the potential risk of being carriers of the mutant gene(s).
In the United States, HCM screening is not routine and, citing cost concerns, the American Heart Association has "consistently opposed" routine screening.
Canada
Canadian genetic testing guidelines and recommendations for individuals diagnosed with HCM are as follows:
For individuals suspected of having HCM:
UK
A post-mortem following the death of popular TV presenter David Frost in 2013 showed he suffered from HCM, though it didn’t contribute to his death and his family wasn’t informed. The sudden cardiac death of his 31-year-old son in 2015 led the family to collaborate with the British Heart Foundation to raise funds for better screening.
Diagnosis
A diagnosis of hypertrophic cardiomyopathy is based upon a number of features of the disease process. While there is use of echocardiography, cardiac catheterization, or cardiac MRI in the diagnosis of the disease, other important considerations include ECG, genetic testing (although not primarily used for diagnosis), and any family history of HCM or unexplained sudden death in otherwise healthy individuals.
Obstructive or non-obstructive
Depending on whether the distortion of normal heart anatomy causes an obstruction of the outflow of blood from the left ventricle of the heart, HCM can be classified as obstructive or non-obstructive.
Physical examination
The physical findings of HCM are associated with the dynamic outflow obstruction that is often present with this disease.
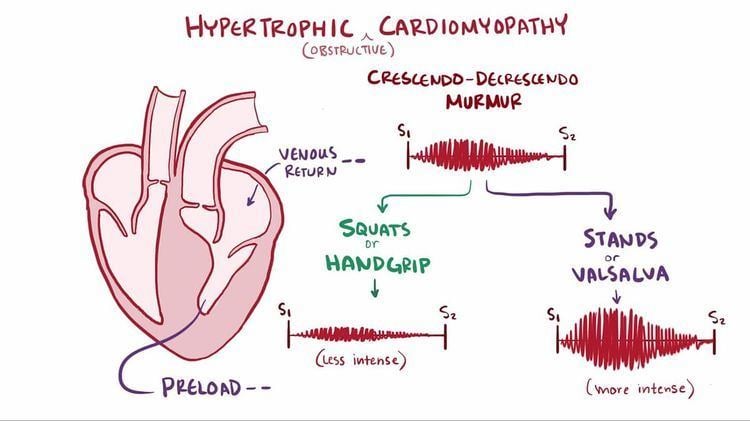
Upon auscultation, the heart murmur will sound similar to the murmur of aortic stenosis. However, a murmur due to HCM will increase in intensity with any maneuver that decreases the volume of blood in the left ventricle (such as standing abruptly or the strain phase of a Valsalva maneuver). Administration of amyl nitrite will also accentuate the murmur by decreasing venous return to the heart. Classically, the murmur is also loudest at the left parasternal edge, 4th intercostal space, rather than in the aortic area.
If dynamic outflow obstruction exists, physical examination findings that can be elicited include the pulsus bisferiens and the double apical impulse with each ventricular contraction. These findings, when present, can help differentiate HCM from aortic stenosis. In addition, if the individual has premature ventricular contractions (PVCs), the change in the carotid pulse intensity in the beat after the PVC can help differentiate HCM from aortic stenosis. In individuals with HCM, the pulse pressure will decrease in the beat after the PVC (Brockenbrough–Braunwald–Morrow sign), while in aortic stenosis, the pulse pressure will increase. However, post-PVC left ventricular systolic pressure and murmur intensity increase in both aortic stenosis as well as HCM.
Cardiac catheterization
Upon cardiac catheterization, catheters can be placed in the left ventricle and the ascending aorta, to measure the pressure difference between these structures. In normal individuals, during ventricular systole, the pressure in the ascending aorta and the left ventricle will equalize, and the aortic valve is open. In individuals with aortic stenosis or with HCM with an outflow tract gradient, there will be a pressure gradient (difference) between the left ventricle and the aorta, with the left ventricular pressure higher than the aortic pressure. This gradient represents the degree of obstruction that has to be overcome in order to eject blood from the left ventricle.
The Brockenbrough–Braunwald–Morrow sign is observed in individuals with HCM with outflow tract gradient. This sign can be used to differentiate HCM from aortic stenosis. In individuals with aortic stenosis, after a premature ventricular contraction (PVC), the following ventricular contraction will be more forceful, and the pressure generated in the left ventricle will be higher. Because of the fixed obstruction that the stenotic aortic valve represents, the post-PVC ascending aortic pressure will increase as well. In individuals with HCM, however, the degree of obstruction will increase more than the force of contraction will increase in the post-PVC beat. The result of this is that the left ventricular pressure increases and the ascending aortic pressure decreases, with an increase in the LVOT gradient.
While the Brockenbrough–Braunwald–Morrow sign is most dramatically demonstrated using simultaneous intra-cardiac and intra-aortic catheters, it can be seen on routine physical examination as a decrease in the pulse pressure in the post-PVC beat in individuals with HCM.
Asymptomatic patients
A significant number of patients with hypertrophic cardiomyopathy do not have any symptoms and will have normal life expectancies, though they should be counseled to avoid particularly strenuous activities or competitive athletics. These patients should also be screened for risk factors for sudden cardiac death. In patients with resting or inducible outflow obstructions, situations that will cause dehydration or vasodilation (such as the use of vasodilatory or diuretic blood pressure medications) should be avoided. Septal reduction therapy is not recommended in asymptomatic patients.
Medications
The primary goal of medications is to relieve symptoms such as chest pain, shortness of breath, and palpitations. Beta blockers are considered first-line agents, as they can slow down the heart rate and decrease the likelihood of ectopic beats. For patients who cannot tolerate beta blockers or do not have good control of symptoms with beta blockers, nondihydropyridine calcium channel blockers such as verapamil can be used, but are potentially harmful in patients in a setting of low blood pressure or severe shortness of breath at rest. These medications also decrease the heart rate, though their use in patients with severe outflow obstruction, elevated pulmonary artery wedge pressure, and low blood pressures should be done with caution. Dihydropyridine calcium channel blockers should be avoided in patients with evidence of obstruction. For patients whose symptoms are not relieved by the above treatments, disopyramide can be considered for further symptom relief. Diuretics can be considered for patients with evidence of fluid overload, though cautiously used in those with evidence of obstruction. Patients who continue to have symptoms despite drug therapy can consider more invasive therapies. Intravenous phenylephrine (or another pure vasoconstricting agent) can be used in the acute setting of low blood pressure in those with obstructive hypertrophic cardiomyopathy who do not respond to fluid administration.
Surgical septal myectomy
Surgical septal myectomy is an open-heart operation done to relieve symptoms in patients who remain severely symptomatic despite medical therapy. It has been performed successfully since the early 1960s. Surgical septal myectomy uniformly decreases left ventricular outflow tract obstruction and improves symptoms, and in experienced centers has a surgical mortality of less than 1%, as well as 85% success rate. It involves a median sternotomy (general anesthesia, opening the chest, and cardiopulmonary bypass) and removing a portion of the interventricular septum. Surgical myectomy resection that focuses just on the subaortic septum, to increase the size of the outflow tract to reduce Venturi forces, may be inadequate to abolish systolic anterior motion (SAM) of the anterior leaflet of the mitral valve. With this limited resection, the residual mid-septal bulge still redirects flow posteriorly; SAM persists because flow still gets behind the mitral valve. It is only when the deeper portion of the septal bulge is resected that flow is redirected anteriorly away from the mitral valve, abolishing SAM. With this in mind, a modification of the Morrow myectomy termed extended myectomy, mobilization and partial excision of the papillary muscles has become the excision of choice. In selected patients with particularly large redundant mitral valves, anterior leaflet plication may be added to complete separation of the mitral valve and outflow. Complications of septal myectomy surgery include possible death, arrhythmias, infection, incessant bleeding, septal perforation/defect, stroke.
Alcohol septal ablation
Alcohol septal ablation, introduced by Ulrich Sigwart in 1994, is a percutaneous technique that involves injection of alcohol into one or more septal branches of the left anterior descending artery. This is a catheter technique with results similar to the surgical septal myectomy procedure but is less invasive, since it does not involve general anaesthesia and opening of the chest wall and pericardium (which are done in a septal myectomy). In a select population with symptoms secondary to a high outflow tract gradient, alcohol septal ablation can reduce the symptoms of HCM. In addition, older individuals and those with other medical problems, for whom surgical myectomy would pose increased procedural risk, would likely benefit from the less-invasive septal ablation procedure.
When performed properly, an alcohol septal ablation induces a controlled heart attack, in which the portion of the interventricular septum that involves the left ventricular outflow tract is infarcted and will contract into a scar. Which patients are best served by surgical myectomy, alcohol septal ablation, or medical therapy is an important topic and one which is intensely debated in medical scientific circles.
Mitral clip
Since 2013, mitral clips have been implanted via catheter as a new strategy to correct the motion of the mitral valve in selected patients with severe obstructive HCM. The device fastens together the mitral valve leaflets to improve the heart's blood outflow. The mitral clip has not yet established the long-term reliability of septal myectomy or alcohol septal ablation, but HCM specialists are increasingly offering the clip as a less-invasive treatment option.
Implantable pacemaker or defibrillator
The use of a pacemaker has been advocated in a subset of individuals, in order to cause asynchronous contraction of the left ventricle. Since the pacemaker activates the interventricular septum before the left ventricular free wall, the gradient across the left ventricular outflow tract may decrease. This form of treatment has been shown to provide less relief of symptoms and less of a reduction in the left ventricular outflow tract gradient when compared to surgical myectomy. Technological advancements have also led to the development of a dual-chamber pacemaker, which is only turned on when needed (in contrast to a regular pacemaker which provides a constant stimulus). Although the dual-chamber pacemaker has shown to decrease ventricular outflow tract obstruction, experimental trials have found only a few individuals with improved symptoms. Unfortunately, researchers suspect that these reports of improved symptoms are due to a placebo effect.
The procedure includes an incision on the anterolateral area below the clavicle. Two leads are then inserted; one into the right atrium and the other into the right ventricular apex via the subclavian veins. Once in place, they are secured and attached to the generator which will remain inside the patient's fascia, anterior to the pectoral muscle. Complications of this procedure include infection, electrical lead and generator malfunction which will require replacement.
For HCM patients who exhibit one or more of the major risk factors for sudden cardiac death, an implantable cardioverter-defibrillator (ICD) or a combination pacemaker/ICD all-in-one unit may be recommended as an appropriate precaution.
Cardiac transplantation
In cases that are unresponsive to all other forms of treatment, cardiac transplantation is one option. It is also the only treatment available for end-stage heart failure. However, transplantation must occur before the onset of symptoms such as pulmonary vessel hypertension, kidney malfunction, and thromboembolism in order for it to be successful. Studies have indicated a seven-year survival rate of 94% in HCM patients after transplantation.
Prognosis
A systematic review from 2002 concluded that: "Overall, HCM confers an annual mortality rate of about 1%... HCM may be associated with important symptoms and premature death but more frequently with no or relatively mild disability and normal life expectancy."
Children
Even though hypertrophic cardiomyopathy (HCM) may be present early in life and is most likely congenital, it is one of the most-uncommon cardiac malformations encountered in pediatric cardiology, largely because the presentation of symptoms is usually absent, incomplete, or delayed into adulthood. Most of the current information pertaining to HCM arises from studies in adult populations, and the implication of these observations for pediatric population is often uncertain. Nonetheless, recent studies in pediatric cardiology have revealed that HCM accounts for 42% of childhood cardiomyopathies, with an annual incidence rate of 0.47/100,000 in children. Further, in asymptomatic cases, sudden death is considered one of the most-feared complications associated with the disease in select pediatric populations. Consequently, the recommended practice is to screen children of affected individuals throughout childhood to detect cardiac abnormalities at an early stage, in the hope of preventing further complications of the disease.
Generally, the diagnosis of HCM in a pediatric population is made during assessment for murmur, congestive heart failure, physical exhaustion, and genetic testing of children of affected individuals. Specifically, echocardiogram (ECHO) has been used as a definitive noninvasive diagnostic tool in nearly all children. ECHO assesses cardiac ventricular size, wall thickness, systolic and diastolic function, and outflow obstruction. Thus, ECHO has been chosen as an ideal means to detect excessive wall thickening of cardiac muscle in HCM.
For children with HCM, treatment strategies aim to reduce disease symptoms and lower the risk of sudden death. Due to the heterogeneity of the disease, treatment is usually modified according to individual patients' needs. β-blockers improve left ventricular filling and relaxation and thereby improve symptoms. In some pediatric patients, β–blockers (e.g., propranolol) were shown effective to reduce the risk of sudden death. Further, calcium channel blockers (verapamil) and antiarrhythmic drugs may be used as an adjunct therapy to β-blockers in symptomatic children. Nonetheless, further testing is needed to determine their definitive benefits.
Other animals
Feline hypertrophic cardiomyopathy (HCM) is the most common heart disease in domestic cats; the disease process and genetics are believed to be similar to the disease in humans. In Maine Coon cats, HCM has been confirmed as an autosomal dominant inherited trait.Numerous cat breeds have HCM as a problem in the breed. The first genetic mutation (in cardiac myosin binding protein C) responsible for feline HCM was discovered in 2005 in Maine Coon cats. A test for this mutation (A31P) is available. About one-third of Maine Coon cats tested for the mutation are either heterozygous or homozygous for the mutation, although many of the cats that are heterozygous have no overt evidence of the disease on an echocardiogram (low penetrance). Some Maine Coon cats with clinical evidence of hypertrophic cardiomyopathy test negative for this mutation, strongly suggesting that another cause exists in the breed. The cardiac myosin binding protein C mutation identified in Maine Coon cats has not been found in any other breed of cat with HCM, but more recently another myosin binding protein C mutation has been identified in Ragdoll cats with HCM. As in humans, feline HCM is not present at birth but develops over time. It has been identified for the first time in cats as young as 6 months of age and at least as old as 7 years of age.
Clinically, cats with hypertrophic cardiomyopathy commonly have a systolic anterior motion of the mitral valve (see graphic). Cats with severe HCM often develop left heart failure (pulmonary edema; pleural effusion) because of severe diastolic dysfunction of the left ventricle. They may also develop a left atrial thrombus that embolizes, most commonly, to the terminal aorta creating acute pain and rear limb paralysis (see below). Sudden death can also occur but appears to be uncommon.
There is no cure for feline HCM. Many but not all cats have a heart murmur. Many cats that have a heart murmur do not have HCM. Frequently the first signs that a cat has HCM are tachypnea/dyspnea due to heart failure or acute pain and paralysis due to systemic thromboembolism. While medication is commonly given to cats with HCM that have no clinical signs, no medication has been shown to be helpful at this stage and it has been shown that an ACE inhibitor is not beneficial until heart failure is present (at which time a diuretic is most beneficial). Diltiazem generally produces no demonstrable benefit. Atenolol is commonly administered when a severe systolic anterior motion of the mitral valve is present.
Feline arterial thromboembolism (FATE) is a relatively common and devastating complication of feline HCM and other feline cardiomyopathies. The thrombus generally forms in the left atrium, most commonly the left auricle. The formation is thought to be primarily due to blood flow stasis. Classically, the thromboembolism lodges at the iliac trifurcation of the aorta, occluding either one or both of the common iliac arteries. Clinically this presents as a cat with complete loss of function in one or both hind limbs. The hind limbs are cold and the cat is in considerable pain. Emboli may, rarely, lodge in other locations, most commonly the right front limb and the renal arteries.
Clopidogrel (Plavix) is used to try to prevent left atrial thrombus formation in cats with HCM and a large left atrium. The FATCAT study at Purdue University demonstrated that it is superior to aspirin for the prevention of a second thrombus from forming in cats that have already experienced a clot. Thrombolytic agents (e.g., tissue plasminogen activator) have been used with some success to break down an existing aortic thromboembolism, but their cost is high and outcome appears to be no better than giving a cat time (48–72 hours) to break down its own clot. Pain management is extremely important. The prognosis for cats with FATE is often poor as they are likely to have significant HCM already and a recurrent bout of FATE is likely. For this reason, euthanasia is often a valid consideration.
In July 2013, Rigo, a 42-year-old Western lowland gorilla, resident in Melbourne Zoo and father of Mzuri, the first gorilla born by artificial insemination, died unexpectedly as a result of HCM. The condition is not uncommon in male gorillas over the age of 30, and in many cases, there is no sign of the disease until the individual's sudden death.