
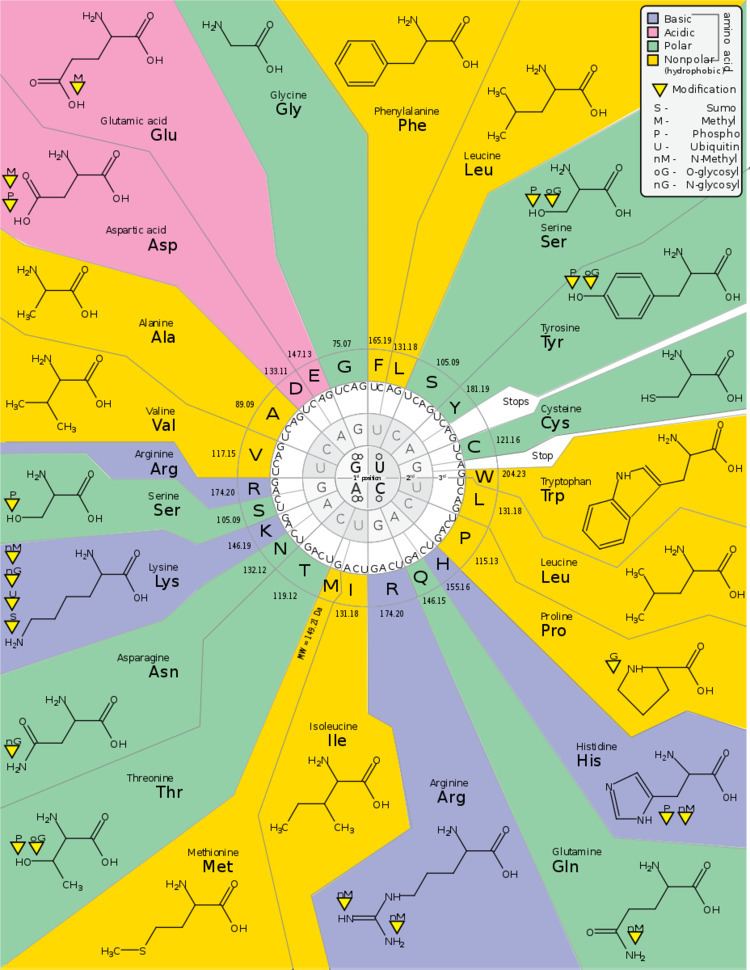
 | ||
The genetic code is the set of rules by which information encoded within genetic material (DNA or mRNA sequences) is translated into proteins by living cells. Translation is accomplished by the ribosome, which links amino acids in an order specified by mRNA, using transfer RNA (tRNA) molecules to carry amino acids and to read the mRNA three nucleotides at a time. The genetic code is highly similar among all organisms and can be expressed in a simple table with 64 entries.
Contents
- Codons
- Expanded genetic codes synthetic biology
- Reading frame
- Startstop codons
- Effect of mutations
- Degeneracy
- Codon usage bias
- RNA codon table
- DNA codon table
- Non standard amino acids
- Variations
- Origin
- References
The code defines how sequences of nucleotide triplets, called codons, specify which amino acid will be added next during protein synthesis. With some exceptions, a three-nucleotide codon in a nucleic acid sequence specifies a single amino acid. The vast majority of genes are encoded with a single scheme (see the RNA codon table). That scheme is often referred to as the canonical or standard genetic code, or simply the genetic code, though variant codes (such as in human mitochondria) exist.
While the "genetic code" determines a protein's amino acid sequence, other genomic regions determine when and where these proteins are produced according to various "gene regulatory codes".
Efforts to understand how proteins are encoded began after DNA's structure was discovered in 1953. George Gamow postulated that sets of three bases must be employed to encode the 20 standard amino acids used by living cells to build proteins, which would allow a maximum of 43 = 64 amino acids.
Codons
The Crick, Brenner et al. experiment first demonstrated that codons consist of three DNA bases. Marshall Nirenberg and Heinrich J. Matthaei were the first to reveal the nature of a codon in 1961.
They used a cell-free system to translate a poly-uracil RNA sequence (i.e., UUUUU...) and discovered that the polypeptide that they had synthesized consisted of only the amino acid phenylalanine. They thereby deduced that the codon UUU specified the amino acid phenylalanine.
This was followed by experiments in Severo Ochoa's laboratory that demonstrated that the poly-adenine RNA sequence (AAAAA...) coded for the polypeptide poly-lysine and that the poly-cytosine RNA sequence (CCCCC...) coded for the polypeptide poly-proline. Therefore, the codon AAA specified the amino acid lysine, and the codon CCC specified the amino acid proline. Using various copolymers most of the remaining codons were then determined.
Subsequent work by Har Gobind Khorana identified the rest of the genetic code. Shortly thereafter, Robert W. Holley determined the structure of transfer RNA (tRNA), the adapter molecule that facilitates the process of translating RNA into protein. This work was based upon Ochoa's earlier studies, yielding the latter the Nobel Prize in Physiology or Medicine in 1959 for work on the enzymology of RNA synthesis.
Extending this work, Nirenberg and Philip Leder revealed the code's triplet nature and deciphered its codons. In these experiments, various combinations of mRNA were passed through a filter that contained ribosomes, the components of cells that translate RNA into protein. Unique triplets promoted the binding of specific tRNAs to the ribosome. Leder and Nirenberg were able to determine the sequences of 54 out of 64 codons in their experiments. Khorana, Holley and Nirenberg received the 1968 Nobel for their work.
The three stop codons were named by discoverers Richard Epstein and Charles Steinberg. "Amber" was named after their friend Harris Bernstein, whose last name means "amber" in German. The other two stop codons were named "ochre" and "opal" in order to keep the "color names" theme.
Expanded genetic codes (synthetic biology)
Since 2001, 40 non-natural amino acids have been added into protein by creating a unique codon (recoding) and a corresponding transfer-RNA:aminoacyl – tRNA-synthetase pair to encode it with diverse physicochemical and biological properties in order to be used as a tool to exploring protein structure and function or to create novel or enhanced proteins.
H. Murakami and M. Sisido extended some codons to have four and five bases. Steven A. Benner constructed a functional 65th (in vivo) codon.
In 2016 the first stable semisynthetic organism was created. It was a (single cell) bacterium with two synthetic bases (called X and Y). The bases survived cell division.
In 2017 a mouse engineered with an extended genetic code that can produce proteins with unnatural amino acids was reported.
Reading frame
A codon is defined by the initial nucleotide from which translation starts and sets the frame for a run of successive triplets, which is known as an "open reading frame" (ORF). For example, the string GGGAAACCC, if read from the first position, contains the codons GGG, AAA, and CCC; if read from the second position, it contains the codons GGA and AAC; and if read from the third position, GAA and ACC. Every sequence can, thus, be read in its 5' → 3' direction in three reading frames, each producing a possibly distinct amino acid sequence (in the given example, Gly-Lys-Pro, Gly-Asn, or Glu-Thr, respectively). DNA is double-stranded defining six possible reading frames, three in the forward orientation on one strand and three reverse on the opposite strand. Protein-coding frames are defined by a start codon, usually the first AUG codon in the sequence.
In eukaryotes, ORFs in exons are often interrupted by introns.
Start/stop codons
Translation starts with a chain-initiation codon or start codon. The start codon alone is not sufficient to begin the process. Nearby sequences such as the Shine-Dalgarno sequence in E. coli and initiation factors are also required to start translation. The most common start codon is AUG, which is read as methionine or, in bacteria, as formylmethionine. Alternative start codons depending on the organism include "GUG" or "UUG"; these codons normally represent valine and leucine, respectively, but as start codons they are translated as methionine or formylmethionine.
The three stop codons have names: UAG is amber, UGA is opal (sometimes also called umber), and UAA is ochre. Stop codons are also called "termination" or "nonsense" codons. They signal release of the nascent polypeptide from the ribosome because no cognate tRNA has anticodons complementary to these stop signals, allowing a release factor to bind to the ribosome instead.
Effect of mutations
During the process of DNA replication, errors occasionally occur in the polymerization of the second strand. These errors, mutations, can affect an organism's phenotype, especially if they occur within the protein coding sequence of a gene. Error rates are typically 1 error in every 10–100 million bases—due to the "proofreading" ability of DNA polymerases.
Missense mutations and nonsense mutations are examples of point mutations that can cause genetic diseases such as sickle-cell disease and thalassemia respectively. Clinically important missense mutations generally change the properties of the coded amino acid residue among basic, acidic, polar or non-polar states, whereas nonsense mutations result in a stop codon.
Mutations that disrupt the reading frame sequence by indels (insertions or deletions) of a non-multiple of 3 nucleotide bases are known as frameshift mutations. These mutations usually result in a completely different translation from the original, and likely cause a stop codon to be read, which truncates the protein. These mutations may impair the protein's function and are thus rare in in vivo protein-coding sequences. One reason inheritance of frameshift mutations is rare is that, if the protein being translated is essential for growth under the selective pressures the organism faces, absence of a functional protein may cause death before the organism becomes viable. Frameshift mutations may result in severe genetic diseases such as Tay-Sachs disease.
Although most mutations that change protein sequences are harmful or neutral, some mutations have benefits. These mutations may enable the mutant organism to withstand particular environmental stresses better than wild type organisms, or reproduce more quickly. In these cases a mutation will tend to become more common in a population through natural selection. Viruses that use RNA as their genetic material have rapid mutation rates, which can be an advantage, since these viruses thereby evolve rapidly, and thus evade the immune system defensive responses. In large populations of asexually reproducing organisms, for example, E. coli, multiple beneficial mutations may co-occur. This phenomenon is called clonal interference and causes competition among the mutations.
Degeneracy
Degeneracy is the redundancy of the genetic code. This term was given by Bernfield and Nirenberg. The genetic code has redundancy but no ambiguity (see the codon tables below for the full correlation). For example, although codons GAA and GAG both specify glutamic acid (redundancy), neither specifies another amino acid (no ambiguity). The codons encoding one amino acid may differ in any of their three positions. For example, the amino acid leucine is specified by YUR or CUN (UUA, UUG, CUU, CUC, CUA, or CUG) codons (difference in the first or third position indicated using IUPAC notation), while the amino acid serine is specified by UCN or AGY (UCA, UCG, UCC, UCU, AGU, or AGC) codons (difference in the first, second, or third position). A practical consequence of redundancy is that errors in the third position of the triplet codon cause only a silent mutation or an error that would not affect the protein because the hydrophilicity or hydrophobicity is maintained by equivalent substitution of amino acids; for example, a codon of NUN (where N = any nucleotide) tends to code for hydrophobic amino acids. NCN yields amino acid residues that are small in size and moderate in hydropathy; NAN encodes average size hydrophilic residues. The genetic code is so well-structured for hydropathy that a mathematical analysis (Singular Value Decomposition) of 12 variables (4 nucleotides x 3 positions) yields a remarkable correlation (C = 0.95) for predicting the hydropathy of the encoded amino acid directly from the triplet nucleotide sequence, without translation. Note in the table, below, eight amino acids are not affected at all by mutations at the third position of the codon, whereas in the figure above, a mutation at the second position is likely to cause a radical change in the physicochemical properties of the encoded amino acid.
Codon usage bias
The frequency of codons, also known as codon usage bias, can vary from species to species with functional implications for the control of translation. The following codon usage table is for the human genome.
A Amino acid. B Fraction of each codon among all those specifying a given amino acid. C Frequency among 40,662,582 codons of 93,487 coding sequences. D Number.
RNA codon table
A The codon AUG both codes for methionine and serves as an initiation site: the first AUG in an mRNA's coding region is where translation into protein begins.B ^ ^ ^ The historical basis for designating the stop codons as amber, ochre and opal is described in an autobiography by Sydney Brenner and in a historical article by Bob Edgar.DNA codon table
The DNA codon table is essentially identical to that for RNA, but with U replaced by T.
Non-standard amino acids
In some proteins, non-standard amino acids are substituted for standard stop codons, depending on associated signal sequences in the messenger RNA. For example, UGA can code for selenocysteine and UAG can code for pyrrolysine. Selenocysteine became to be seen as the 21st amino acid, and pyrrolysine as the 22nd. Unlike selenocysteine, pyrrolysine-encoded UAG is translated with the participation of a dedicated aminoacyl-tRNA synthetase. Both selenocysteine and pyrrolysine may be present in the same organism. Although the genetic code is normally fixed in an organism, the achaeal prokaryote Acetohalobium arabaticum can expand its genetic code from 20 to 21 amino acids (by including pyrrolysine) under different conditions of growth.
Variations
Variations on the standard code were predicted in the 1970s. The first was discovered in 1979, by researchers studying human mitochondrial genes. Many slight variants were discovered thereafter, including various alternative mitochondrial codes. Small variants such as translation of the codon UGA as tryptophan in Mycoplasma species, and translation of CUG as a serine rather than leucine in yeasts of the "CTG clade" (such as Candida albicans). Because viruses must use the same genetic code as their hosts, modifications to the standard genetic code could interfere with viral protein synthesis or functioning. However, viruses such as totiviruses adapted to the host's genetic code modification. In bacteria and archaea, GUG and UUG are common start codon. In rare cases, certain proteins may use alternative start codons.
Variant genetic codes used by an organism can be inferred by identifying highly conserved genes encoded in that genome, and comparing its codon usage to the amino acids in homologous proteins of other organisms. For example, the program FACIL infers a genetic code by searching which amino acids in homologous protein domains are most often aligned to every codon. The resulting amino acid probabilities for each codon are displayed in a genetic code logo, that also shows the support for a stop codon.
Despite these differences, all known naturally occurring codes are very similar. The coding mechanism is the same for all organisms: three-base codons, tRNA, ribosomes, single direction reading and translating single codons into single amino acids.
Origin
The genetic code is a key part of the story of life. The main hypothesis for life's origin is the RNA world hypothesis. Any model for the emergence of genetic code is intimately related to a model of the transfer from ribozymes (RNA enzymes) to proteins as the principal enzymes in cells. In line with the RNA world hypothesis, transfer RNA molecules appear to have evolved before modern aminoacyl-tRNA synthetases, so the latter cannot be part of the explanation of its patterns.
A hypothetical randomly evolved genetic code further motivates a biochemical or evolutionary model for its origin. If amino acids were randomly assigned to triplet codons, there would be 1.5 × 1084 possible genetic codes. This number is found by calculating the number of ways that 21 items (20 amino acids plus one stop) can be placed in 64 bins, wherein each item is used at least once. However, the distribution of codon assignments in the genetic code is nonrandom. In particular, the genetic code clusters certain amino acid assignments.
Amino acids that share the same biosynthetic pathway tend to have the same first base in their codons. This could be an evolutionary relic of an early, simpler genetic code with fewer amino acids that later evolved to code a larger set of amino acids. It could also reflect steric and chemical properties that had another effect on the codon during its evolution. Amino acids with similar physical properties also tend to have similar codons, reducing the problems caused by point mutations and mistranslations.
Given the non-random genetic triplet coding scheme, a tenable hypothesis for the origin of genetic code could address multiple aspects of the codon table, such as absence of codons for D-amino acids, secondary codon patterns for some amino acids, confinement of synonymous positions to third position, the small set of only 20 amino acids (instead of a number approaching 64), and the relation of stop codon patterns to amino acid coding patterns.
Three main hypotheses address the origin of the genetic code. Many models belong to one of them or to a hybrid:
Hypotheses have addressed a variety of scenarios: