
Specialty medical genetics ICD-9-CM 757.33 DiseasesDB 14198 | ICD-10 Q82.1 OMIM 278700 MedlinePlus 001467 | |
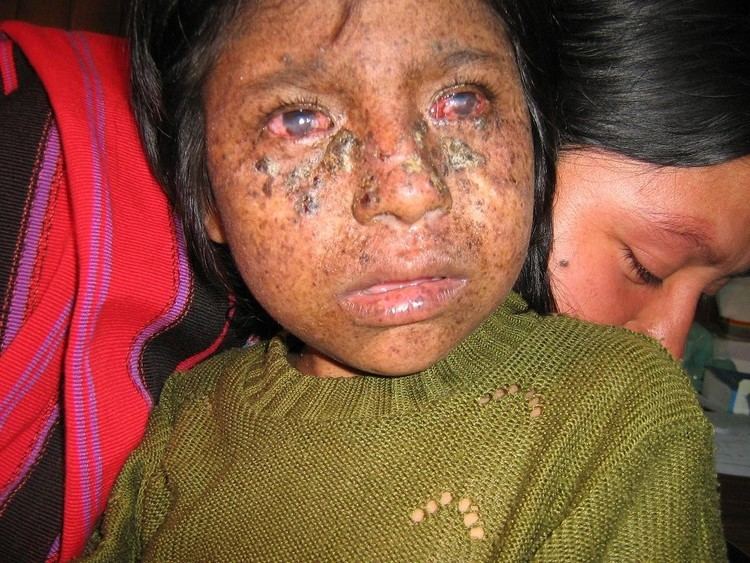 | ||
Xeroderma pigmentosum (XP) is a rare autosomal recessive genetic disorder of DNA repair in which the ability to repair damage caused by ultraviolet (UV) light is deficient. In extreme cases, all exposure to sunlight must be forbidden, no matter how small; as such, individuals with the disease are often colloquially referred to as "Moon child". Multiple basal cell carcinomas (basaliomas) and other skin malignancies frequently occur at a young age in those with XP; metastatic malignant melanoma and squamous cell carcinoma are the two most common causes of death in XP victims. This disease is present in both genders and in all races, with an incidence of 1:250,000 in the United States. XP is roughly six times more common in Japanese people than in other groups.
Contents
Normally, damage to DNA in epidermal cells occurs during exposure to UV light. The absorption of the high-energy light leads to the formation of pyrimidine dimers, namely cyclobutane-pyrimidine dimers and pyrimidine-6-4-pyrimidone photoproducts. In a healthy, normal human being, the damage is first excised by endonucleases. DNA polymerase then repairs the missing sequence, and ligase "seals" the transaction. This process is known as nucleotide excision repair.
Genetics
One of the most frequent defects in xeroderma pigmentosum is an autosomal recessive genetic defect in which nucleotide excision repair (NER) enzymes are mutated, leading to a reduction in or elimination of NER. If left unchecked, damage caused by ultraviolet light can cause mutations in individual cell's DNA. The causes of the neurological abnormalities are poorly understood and are not connected with exposure to ultraviolet light. The most current theories suggest that oxidative DNA damage is generated during normal metabolism in the central nervous system, and that some types of this damage must be repaired by NER
Since DNA repair is under genetic control, it can easily undergo mutations. Many genetic disorders such as xeroderma pigmentosum (XP; MIM 278700) are caused by mutations in genes that repair DNA. If the gene was not repaired correctly it could cause xeroderma pigmentosum in individuals. The autosomal recessive disorder xeroderma pigmentosum or XP has a frequency of 1 in every 250,000 individuals of all races and ethnic groups. Those affected with the autosomal recessive disorder XP are extremely sensitive to UV light produced by the sun and even with a short exposure to it causes dry, flaking skin and pigmented spots that can develop into skin cancer. Individuals with XP are about 1,000 times more likely to develop skin cancer than individuals without the disorder.
The molecular defects in XP cells result in a greatly elevated induction of mutations in sun-exposed skin of affected individuals. This increased mutation frequency probably accounts for the pigmentation changes and the skin cancers. Examination of mutations in the p53 gene in tumors from XP patients reveal p53 mutations characteristic of UV exposure in the majority of tumors As with all genetic disorders, genetic counseling and psychological support is appropriate for the families, to discuss probability of occurrence in future pregnancies, feelings of isolation and concern about career prospects. Although there is no cure for xeroderma pigmentosum, the effects can be minimized by getting protection from the sunlight and if possible early removal of precancerous lesions. The most common fate for individuals with XP is early death from cancer due to the fact that they need to take outstanding measures to protect themselves from the dangers of the UV light. But if there is an absence of neurological problems and the individual is always protected or away from sunlight, the prognosis is good.
Types
There are seven complementation groups, plus one variant form:
Symptoms
Symptoms include:
Treatment
The most obvious, and often important part of treatment, is avoiding exposure to sunlight. This includes wearing protective clothing and using sunscreen (physical and chemical). Keratosis can also be treated using cryotherapy or fluorouracil.
Prognosis
Fewer than 40% of individuals with the disease survive beyond the age of 20. Some XP victims with less severe cases do manage to live well into their 40s.
Functions of XP repair proteins
The XPA protein acts during NER as a scaffold for assembly of other DNA repair proteins at sites of DNA damage to ensure appropriate excision of the damage.
The XPB(ERCC3) protein is employed in unwinding the DNA double helix after DNA damage is initially recognized. Mutations in the XPB(ERCC3) gene can lead to XP or XP combined with Cockayne syndrome.
The XPC protein forms a complex with RAD23B protein to form the initial damage recognition factor in global genomic nucleotide excision repair (GG-NER). This complex recognizes a wide variety of damages that thermodynamically destabilize DNA duplexes.
The XPD(ERCC2) protein, in combination with the XPB helicase-containing transcription/repair complex TFIIH, is employed in unwinding the DNA duplex after damage is initially recognized. Mutations in the XPD(ERCC2) gene cause a variety of syndromes; XP, trichothiodystrophy (TTD), or a combination of XP and Cockayne syndrome (XPCS). Both trichothiodystrophy and Cockayne syndrome display features of premature aging, suggesting an association between deficient DNA repair and premature aging (see DNA damage theory of aging).
XPE is a heterodimeric protein composed of two subunits. The larger subunit DDB1 primarily functions as a core component of CUL4A- and CUL4B-based E3 ubiquitin ligase complexes. Substrates that are ubiquitinnated by these complexes include proteins employed in DNA repair.
The XPF(ERCC4) protein together with the ERCC1 protein forms a complex usually designated ERCC1-XPF. This complex separates the DNA helix for a short distance on either side of the site of damage. It then acts as a endonuclease to incise the damaged DNA strand on the 5’ side of the damaged site. Mutant cells with deficient ERCC1-XPF are not only defective in NER, but also in the repair of double-strand breaks and inter-strand crosslinks.
The XPG protein is an endonuclease that incises DNA during NER at the 3’ side of the damaged nucleotide. Mutations in the XPG(ERCC5) gene can lead to XP alone, or in combination with Cockayne syndrome (CS), or in combination with infantile lethal cerebro-oculo-facio-skeletal syndrome.
In popular culture
These fictional characters have XP: