
ICD-10 Q85.1 OMIM 191100 613254 | ICD-9-CM 759.5 DiseasesDB 13433 | |
 | ||
Synonyms Tuberous sclerosis complex (TSC),Bourneville disease Specialty neurology, medical genetics | ||
Tuberous sclerosis complex (TSC) is a rare multisystem genetic disease that causes benign tumors to grow in the brain and on other vital organs such as the kidneys, heart, liver, eyes, lungs, and skin. A combination of symptoms may include seizures, intellectual disability, developmental delay, behavioral problems, skin abnormalities, and lung and kidney disease. TSC is caused by a mutation of either of two genes, TSC1 and TSC2, which code for the proteins hamartin and tuberin, respectively. These proteins act as tumor growth suppressors, agents that regulate cell proliferation and differentiation.
Contents
- Signs and symptoms
- Central nervous system
- Kidneys
- Lungs
- Heart
- Skin
- Eyes
- Pancreas
- Variability
- Genetics
- Pathophysiology
- Diagnosis
- Management
- Prognosis
- Epidemiology
- History
- References
The name, composed of the Latin tuber (swelling) and the Greek skleros (hard), refers to the pathological finding of thick, firm, and pale gyri, called "tubers", in the brains of patients post mortem. These tubers were first described by Désiré-Magloire Bourneville in 1880; the cortical manifestations may sometimes still be known by the eponym Bourneville's disease (English /bɔərnˈviːl/) or Bourneville–Pringle disease (after Bourneville and John James Pringle). The full name of "tuberous sclerosis complex" is preferred among medical professionals and researchers today because the disease has manifestations outside of the brain. Furthermore, the name reduces confusion with tuberculosis, a more well-known disease.
Signs and symptoms
The physical manifestations of TSC are due to the formation of hamartia (malformed tissue such as the cortical tubers), hamartomas (benign growths such as facial angiofibroma and subependymal nodules), and very rarely, cancerous hamartoblastomas. The effect of these on the brain leads to neurological symptoms such as seizures, intellectual disability, developmental delay, and behavioral problems. Symptoms also include trouble in school and concentration problems.
Central nervous system
About 50% of people with TSC have learning difficulties ranging from mild to significant, and studies have reported that between 25% and 61% of affected individuals meet the diagnostic criteria for autism, with an even higher proportion showing features of a broader pervasive developmental disorder. A 2008 study reported self-injurious behavior in 10% of people with TSC. Other behaviors and disabilities, such as ADHD, aggression, behavioral outbursts, and OCD can also occur. Lower IQ is associated with more brain involvement on MRI.
Three types of brain tumours may be associated with TSC:
Classic intracranial manifestations of TSC include subependymal nodules and cortical/subcortical tubers.
The tubers are typically triangular in configuration, with the apex pointed towards the ventricles, and are thought to represent foci of abnormal neuronal migration. The T2 signal abnormalities may subside in adulthood, but will still be visible on histopathological analysis. On magnetic resonance imaging, TSC patients can exhibit other signs consistent with abnormal neuron migration such as radial white matter tracts hyperintense on T2WI and heterotopic gray matter.
Subependymal nodules are composed of abnormal, swollen glial cells and bizarre multinucleated cells which are indeterminate for glial or neuronal origin. Interposed neural tissue is not present. These nodules have a tendency to calcify as the patient ages. A nodule that markedly enhances and enlarges over time should be considered suspicious for transformation into a subependymal giant cell astrocytoma, which typically develops in the region of the foramen of Monro, in which case it is at risk of developing an obstructive hydrocephalus.
A variable degree of ventricular enlargement is seen, either obstructive (e.g. by a subependymal nodule in the region of the foramen of Monro) or idiopathic in nature.
Kidneys
Between 60 and 80% of TSC patients have benign tumors (once thought hamartomatous, but now considered true neoplasms) of the kidneys called angiomyolipomas frequently causing hematuria. These tumors are composed of vascular (angio–), smooth muscle (–myo–), and fat (–lip-) tissue. Although benign, an angiomyolipoma larger than 4 cm is at risk for a potentially catastrophic hemorrhage either spontaneously or with minimal trauma. Angiomyolipomas are found in about one in 300 people without TSC. However, those are usually solitary, whereas in TSC they are commonly multiple and bilateral.
About 20-30% of people with TSC have renal cysts, causing few problems. However, 2% may also have autosomal dominant polycystic kidney disease.
Very rare (< 1%) problems include renal cell carcinoma and oncocytomas (benign adenomatous hamartoma).
Lungs
Patients with TSC can develop progressive replacement of the lung parenchyma with multiple cysts. This process is identical to another disease called lymphangioleiomyomatosis (LAM). Recent genetic analysis has shown that the proliferative bronchiolar smooth muscle in TSC-related lymphangioleiomyomatosis is monoclonal metastasis from a coexisting renal angiomyolipoma. Cases of TSC-related lymphangioleiomyomatosis recurring following lung transplant have been reported.
Heart
Rhabdomyomas are benign tumors of striated muscle. A cardiac rhabdomyoma can be discovered using echocardiography in around 50% of people with TSC. However, the incidence in the newborn may be as high as 90% and in adults as low as 20%. These tumors grow during the second half of pregnancy and regress after birth. Many disappear entirely; alternatively, the tumor size remains constant as the heart grows, which has much the same effect.
Problems due to rhabdomyomas include obstruction, arrhythmia, and a murmur. Such complications occur almost exclusively during pregnancy or within the child's first year. Prenatal ultrasound, performed by an obstetric sonographer specializing in cardiology, can detect a rhabdomyoma after 20 weeks. This rare tumour is a strong indicator of TSC in the child, especially if a family history of TSC exists.
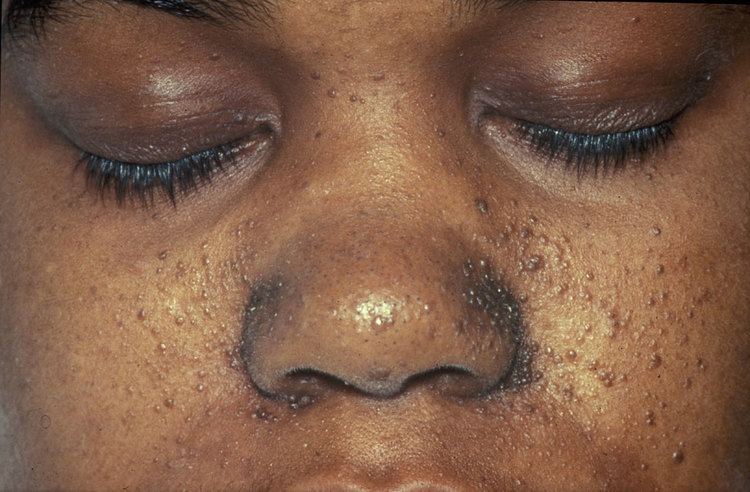
Skin
Some form of dermatological sign is present in 96% of individuals with TSC. Most cause no problems, but are helpful in diagnosis. Some cases may cause disfigurement, necessitating treatment. The most common skin abnormalities include:
Eyes
Retinal lesions, called astrocytic hamartomas (or "phakomas"), which appear as a greyish or yellowish-white lesion in the back of the globe on the ophthalmic examination. Astrocytic hamartomas can calcify, and they are in the differential diagnosis of a calcified globe mass on a CT scan.
Nonretinal lesions associated with TSC include:
Pancreas
Pancreatic neuroendocrine tumours have been described in rare cases of TSC.
Variability
Individuals with TSC may experience none or all of the clinical signs discussed above. The following table shows the prevalence of some of the clinical signs in individuals diagnosed with TSC.
Genetics
TSC is a genetic disorder with an autosomal dominant pattern of inheritance, variable expressivity, and complete penetrance. Two-thirds of TSC cases result from sporadic genetic mutations, not inheritance, but their offspring may inherit it from them. Current genetic tests have difficulty locating the mutation in roughly 20% of individuals diagnosed with the disease. So far, it has been mapped to two genetic loci, TSC1 and TSC2.
TSC1 encodes for the protein hamartin, is located on chromosome 9 q34, and was discovered in 1997. TSC2 encodes for the protein tuberin, is located on chromosome 16 p13.3, and was discovered in 1993. TSC2 is contiguous with PKD1, the gene involved in one form of polycystic kidney disease (PKD). Gross deletions affecting both genes may account for the 2% of individuals with TSC who also develop polycystic kidney disease in childhood. TSC2 has been associated with a more severe form of TSC. However, the difference is subtle and cannot be used to identify the mutation clinically. Estimates of the proportion of TSC caused by TSC2 range from 55% to 90%.
TSC1 and TSC2 are both tumor suppressor genes that function according to Knudson's "two hit" hypothesis. That is, a second random mutation must occur before a tumor can develop. This explains why, despite its 100% penetrance, TSC has wide expressivity.
Pathophysiology
Hamartin and tuberin function as a complex which is involved in the control of cell growth and cell division. The complex appears to interact with RHEB GTPase, thus sequestering it from activating mTOR signalling, part of the growth factor (insulin) signalling pathway. Thus, mutations at the TSC1 and TSC2 loci result in a loss of control of cell growth and cell division, and therefore a predisposition to forming tumors. TSC affects tissues from different germ layers. Cutaneous and visceral lesions may occur, including adenoma sebaceum, cardiac rhabdomyomas, and renal angiomyolipomas. The central nervous system lesions seen in this disorder include hamartomas of the cortex, hamartomas of the ventricular walls, and subependymal giant cell tumors, which typically develop in the vicinity of the foramina of Monro.
Molecular genetic studies have defined at least two loci for TSC. In TSC1, the abnormality is localized on chromosome 9q34, but the nature of the gene protein, called hamartin, remains unclear. No missense mutations occur in TSC1. In TSC2, the gene abnormalities are on chromosome 16p13. This gene encodes tuberin, a guanosine triphosphatase–activating protein. The specific function of this protein is unknown. In TSC2, all types of mutations have been reported; new mutations occur frequently. Few differences have yet been observed in the clinical phenotypes of patients with mutation of one gene or the other.
Diagnosis
No pathognomonic clinical signs for TSC complex are seen. Many signs are present in individuals who are healthy (although rarely), or who have another disease. In order to meet diagnostic criteria for TSC complex, an individual must either have: 1) Two or more major criteria; or 2) One major criterion along with two or more minor criteria.
In infants, the first clue is often the presence of seizures, delayed development, or white patches on the skin. A full clinical diagnosis involves:
The various signs are then marked against the diagnostic criteria to produce a level of diagnostic certainty:
Due to the wide variety of mutations leading to TSC, no simple genetic tests are available to identify new cases, nor are any biochemical markers known for the gene defects. However, once a person has been clinically diagnosed, the genetic mutation can usually be found. The search is time-consuming and has a 15% failure rate, which is thought to be due to somatic mosaicism. If successful, this information can be used to identify affected family members, including prenatal diagnosis. As of 2006, preimplantation diagnosis is not widely available.
Management
TSC typically affects multiple organ systems and manifests differently in each patient and in different stages of the life course. Drug therapy, surgery, and other interventions can be effective in managing some of the manifestations and symptoms of TSC.
In the United States, the Food and Drug Administration has approved several drugs for managing some of the major manifestations of TSC. The antiepileptic medication vigabatrin was approved in 2009 for treatment of infantile spasms and was recommended as first-line therapy for infantile spasms in children with TSC by the 2012 International TSC Consensus Conference. Adrenocorticotropic hormone was approved in 2010 to treat infantile spasms. Everolimus was approved for treatment of TSC-related tumors in the brain (subependymal giant cell astrocytoma) in 2010 and in the kidneys (renal angiomyolipoma) in 2012. Everolimus also showed evidence of effectiveness at treating epilepsy in some people with TSC. In 2017, the European Commission approved everolimus for treatment of refractory partial-onset seizures associated with TSC.
Neurosurgical intervention may reduce the severity and frequency of seizures in TSC patients. Embolization and other surgical interventions can be used to treat renal angiomyolipoma with acute hemorrhage. Surgical treatments for symptoms of lymphangioleiomyomatosis (LAM) in adult TSC patients include pleurodesis to prevent pneumothorax and lung transplantation in the case of irreversible lung failure.
Other treatments that have been used to treat TSC manifestations and symptoms include a ketogenic diet for intractable epilepsy and pulmonary rehabilitation for LAM.
Prognosis
The prognosis for individuals with TSC depends on the severity of symptoms, which range from mild skin abnormalities to varying degrees of learning disabilities and epilepsy to severe intellectual disability, uncontrollable seizures, and kidney failure. Those individuals with mild symptoms generally do well and live long, productive lives, while individuals with the more severe form may have serious disabilities. However, with appropriate medical care, most individuals with the disorder can look forward to normal life expectancy.
A study of 30 TSC patients in Egypt found, "...earlier age of seizures commencement (<6 months) is associated with poor seizure outcome and poor intellectual capabilities. Infantile spasms and severely epileptogenic EEG patterns are related to the poor seizure outcome, poor intellectual capabilities and autistic behavior. Higher tubers numbers is associated with poor seizure outcome and autistic behavior. Left-sided tuber burden is associated with poor intellect, while frontal location is more encountered in ASD. So, close follow up for the mental development and early control of seizures are recommended in a trial to reduce the risk factors of poor outcome. Also early diagnosis of autism will allow for earlier treatment and the potential for better outcome for children with TSC."
Leading causes of death include renal disease, brain tumour, lymphangioleiomyomatosis of the lung, and status epilepticus or bronchopneumonia in those with severe mental handicap. Cardiac failure due to rhabdomyomas is a risk in the fetus or neonate, but is rarely a problem subsequently. Kidney complications such as angiomyolipoma and cysts are common, and more frequent in females than males and in TSC2 than TSC1. Renal cell carcinoma is uncommon. Lymphangioleiomyomatosis is only a risk for females with angiomyolipomas. In the brain, the subependymal nodules occasionally degenerate to subependymal giant cell astrocytomas. These may block the circulation of cerebrospinal fluid around the brain, leading to hydrocephalus.
Detection of the disease should be followed by genetic counselling. It is also important to realise that though the disease does not have a cure, symptoms can be treated symptomatically. Hence, awareness regarding different organ manifestations of TSC is important.
Epidemiology
TSC occurs in all races and ethnic groups, and in both genders. The live-birth prevalence is estimated to be between 10 and 16 cases per 100,000. A 1998 study estimated total population prevalence between about 7 and 12 cases per 100,000, with more than half of these cases undetected. These estimates are significantly higher than those produced by older studies, when TSC was regarded as an extremely rare disease. This is due to the invention of CT and ultrasound scanning having enabled the diagnosis of many nonsymptomatic cases. Prior to this, the diagnosis of TSC was largely restricted to severely affected individuals with Vogt's triad of learning disability, seizures, and facial angiofibroma. The total population prevalence estimates have steadily increased from 1:150,000 in 1956, to 1:100,000 in 1968, to 1:70,000 in 1971, to 1:34,200 in 1984, to the present figure of 1:12,500 in 1998. Whilst still regarded as a rare disease, TSC is common when compared to many other genetic diseases.
History
TSC first came to medical attention when dermatologists described the distinctive facial rash (1835 and 1850). A more complete case was presented by von Recklinghausen (1862), who identified heart and brain tumours in a newborn who had only briefly lived. However, Bourneville (1880) is credited with having first characterized the disease, coining the name "tuberous sclerosis", thus earning the eponym Bourneville's disease. The neurologist Vogt (1908) established a diagnostic triad of epilepsy, idiocy, and adenoma sebaceum (an obsolete term for facial angiofibroma).
Symptoms were periodically added to the clinical picture. The disease as presently understood was first fully described by Gomez (1979). The invention of medical ultrasound, CT and MRI has allowed physicians to examine the internal organs of live patients and greatly improved diagnostic ability.
In 2002, treatment with rapamycin was found to be effective at shrinking tumours in animals. This has led to human trials of rapamycin as a drug to treat several of the tumors associated with TSC.