
ICD-10 J84.81 ICD-O 9174/1 DiseasesDB 30755 | ICD-9-CM 516.4 OMIM 606690 eMedicine med/1348 radio/415 | |
 | ||
Lymphangioleiomyomatosis (LAM) is a disease. It is rare, progressive and systemic and typically results in cystic lung destruction. It predominantly affects women, especially during childbearing years.
Contents
- Incidence
- Signs and symptoms
- Pathophysiology
- Genetics
- Pathology
- Chest radiograph
- High resolution computed tomography
- Ventilation perfusion scans
- Positron emission tomography
- Abdominal imaging
- Central nervous system imaging
- Pulmonary function studies
- Diagnosis
- Pneumothorax
- Chylothorax
- Angiomyolipoma
- Lymphangioleiomyoma
- Management other
- Pharmacological treatment
- Pregnancy
- Air travel
- Prognosis
- Epidemiology
- Clinical research
- Patient registry
- In popular culture
- References
Incidence
It occurs in more than 30% of women with tuberous sclerosis complex (TSC-LAM), a heritable syndrome that is associated with seizures, cognitive impairment and benign tumors in multiple tissues. Most LAM patients who present for medical evaluation have the sporadic form of the disease (S-LAM), however, which is not associated with other manifestations of tuberous sclerosis complex.
Mild cystic changes consistent with LAM have been described in 10–15% of men with TSC, but symptomatic LAM in males is rare. Sporadic LAM occurs exclusively in women, with one published exception to date. Both TSC-LAM and S-LAM are associated with mutations in tuberous sclerosis genes.
Lung destruction in LAM is a consequence of diffuse infiltration by neoplastic smooth muscle-like cells that invade all lung structures including the lymphatics, airway walls, blood vessels and interstitial spaces. The consequences of vessel and airway obstruction include chylous fluid accumulations, hemoptysis, airflow obstruction and pneumothorax. The typical disease course displays progressive dyspnea on exertion, spaced by recurrent pneumothoraces and in some patients, chylous pleural effusions or ascites.
Estimates of median survival vary from 10 to 30 years, depending on whether hospital-based or population-based cohorts are studied. Most patients have dyspnea on exertion with daily activities by 10 years after symptom onset. Many patients require supplemental oxygen over that interval. An FDA-approved drug for treatment of LAM, sirolimus, is available for stabilization of lung function decline. Lung transplant remains the last resort for patients with advanced disease.
Signs and symptoms
The average age of onset is the early to mid 30s. Exertional dyspnea and spontaneous pneumothorax have been reported as the initial presentation of the disease in 49% and 46% of patients, respectively.
Diagnosis is typically delayed 5 to 6 years. The condition is often misdiagnosed as asthma or chronic obstructive pulmonary disease. The first pneumothorax precedes the diagnosis of LAM in 82% of patients. The consensus clinical definition of LAM includes multiple symptoms:
Pathophysiology
A variable percentage of cells within the LAM lesion contain mutational inactivation of the Tuberous Sclerosis Complex (TSC1 or TSC2) tumor suppressor genes. TSC1 mutations cause a less severe clinical phenotype than TSC2 mutations. The discovery of TSC1/2 gene function as negative regulator of the mammalian target of rapamycin complex 1 (mTORC1) led to successful use of rapamycin analog sirolimus in clinical trials and FDA approval of sirolimus for treatment of LAM.
TSC1 and TSC2 form a tumor suppressor complex that regulates mammalian target of rapamycin (mTOR) signaling complex by directly controlling the activity of the small GTPase Rheb via the GTPase activating protein (GAP) domain of TSC2. Rheb binds to Raptor and controls the activity of mTOR complex 1 (mTORC1) that directly phosphorylates p70 S6 kinase (S6K1) and 4E-BP1. mTOR forms two physically and functionally distinct multiprotein complexes: the rapamycin-sensitive mTORC1 and the rapamycin-insensitive mTORC2. MTORC1 consists of five proteins including Raptor that positively regulate mTOR activity. MTORC2 consists of six proteins including mTOR and Rictor, which defines the activation level of mTORC2 and modulates the assembly of the actin cytoskeleton through Rho GTPases, and Rac1 is required for mTOR activation. In TSC2-null and human LAM cells, Rho GTPase activity is required for cell adhesion, motility, proliferation and survival. Loss of TSC1/TSC2 in LAM induces uncontrolled LAM cell growth and increases LAM cell viability. Upregulation of STAT1 and STAT3 and autophagy are known mediators of LAM cell viability and survival.
LAM cells behave, in many ways, like metastatic tumor cells. LAM cells appear to arise from an extrapulmonary source and migrate to the lung. Increased LAM cell migration and invasiveness is rescued by TSC2 re-expression. The cellular and molecular mechanisms of neoplastic transformation and lung parenchymal destruction by LAM cells remain unknown. Lung remodeling may be mediated by an imbalance between matrix degrading metalloproteinases (MMPs) and their endogenous inhibitors TIMPs. The invasive cell phenotype in LAM is associated with TIMP-3 downregulation and TSC2-dependent upregulation of MMPs.
Clinical and histopathological evidence demonstrate the lymphatic involvement in LAM. The prevailing hypothesis is that LAM lesions secrete the lymphangiogenic factor VEGF-D, recruit lymphatic endothelial cells (LECs) that form lymphatic vessels and induce lung cysts. VEGF-D serum levels are increased in LAM compared to other cystic lung diseases, including pulmonary Langerhans cell histiocytosis, emphysema, Sjögren's syndrome,or Birt–Hogg–Dubé syndrome. VEGF-D levels correlate with the severity of LAM, evaluated as a measure of CT grade (the abundance of chylous effusions and lymphatic involvement). VEGF-D is a secreted homodimeric glycoprotein and a member of the VEGF family of growth factors, is known for its role in cancer lymphangiogenesis and metastasis. Proteolytic processing of VEGF-D affects cognate binding to VEGFR3. Histopathologically, LAM lesions are surrounded by cells that stain for VEGFR3, the lymphatic vessel endothelial hyaluronan receptor 1 (LYVE-1) and podoplanin. VEGF-D binds to the receptor protein tyrosine kinases VEGFR-2 and VEGFR-349 in humans, and to VEGFR3 in mice. Surprisingly, knock-out of VEGF-D in mice has little effect on lymphatic system development. Nevertheless, during tumorigenesis VEGF-D promotes formation of tumor lymphatic vessels and facilitates metastatic spread of cancer cells. However, little is known about a role of abnormal lymphatics and VEGF-D in LAM etiology and pathogenesis.
Genetics
LAM occurs in two settings: in the disease tuberous sclerosis complex (TSC-LAM) and in a sporadic form, in women who do not have TSC (sporadic LAM). In both settings, genetic evidence indicates that LAM is caused by inactivating or “loss of function” mutations in the TSC1 or TSC2 genes, which were cloned in 1997 and 1993 respectively. The TSC1 gene is located on chromosome 9q34 and the TSC2 gene is located on chromosome 16p13. TSC-LAM occurs in women who have germline mutations in either the TSC1 or the TSC2 gene.
Sporadic LAM is primarily associated with somatic TSC2 gene mutations. Germline and somatic mutations in LAM include many types of mutations spread across the genes, with no clear “hot spots,” including missense changes, in-frame deletions and nonsense mutations. Because of the large size of the genes (together they have more than 60 exons) and because mutations can be located virtually anywhere within the genes, mutation detection is often challenging.
On a cellular basis, LAM cells carry bi-allelic inactivation of the TSC2 genes, consistent with the “two-hit” tumor suppressor gene model. The second hit event in LAM cells is often loss of the chromosomal region containing the wild-type copy of the TSC2 gene; this is referred to as loss of heterozygosity or LOH. LOH can be detected in microdissected LAM cells, in angiomyolipomas and lymph nodes from women with LAM, and in circulating LAM cells (cells in blood and urine).
Angiomyolipomas and pulmonary LAM cells from women with the sporadic form of LAM carry identical mutations in TSC2. This, together with the fact that recurrent LAM after lung transplantation carries the same TSC2 mutations as the original LAM, has led to the "benign metastasis" hypothesis that LAM cells can migrate or metastasize from one site to another.
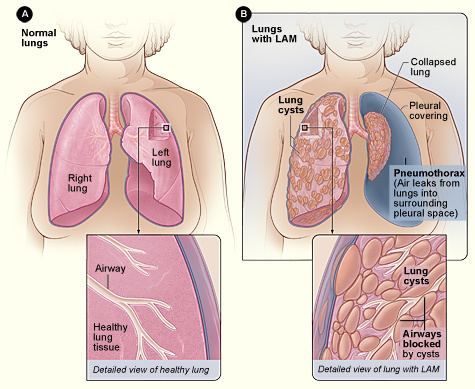
Pathology
Grossly, LAM lungs are enlarged and diffusely cystic, with dilated air spaces as large as several centimeters in diameter. Microscopic examination of the lung reveals foci of smooth muscle-like cell infiltration of the lung parenchyma, airways, lymphatics, and blood vessels associated with areas of thin-walled cystic change. LAM lesions often contain an abundance of lymphatic channels, forming an anastomosing meshwork of slit-like spaces lined by endothelial cells. LAM cells generally expand interstitial spaces without violating tissue planes but have been observed to invade the airways, the pulmonary artery, the diaphragm, aorta, and retroperitoneal fat, to destroy bronchial cartilage and arteriolar walls, and to occlude the lumen of pulmonary arterioles.
There are two major cell morphologies in the LAM lesion: small spindle-shaped cells and cuboidal epithelioid cells. LAM cells stain positively for smooth muscle actin, vimentin, desmin, and, often, estrogen and progesterone receptors. The cuboidal cells within LAM lesions also react with a monoclonal antibody called HMB-45, developed against the premelanosomal protein gp100, an enzyme in the melanogenesis pathway. This immunohistochemical marker is very useful diagnostically, because other smooth muscle–predominant lesions in the lung do not react with the antibody. The spindle-shaped cells of the LAM lesion are more frequently proliferating cell nuclear antigen positive than the cuboidal cells, consistent with a proliferative phenotype. Compared with cigar-shaped normal smooth muscle cells, spindle-shaped LAM cells contain less abundant cytoplasm and are less eosinophilic. Estrogen and progesterone receptors are also present in LAM lesions, but not in adjacent normal lung tissue. LAM lesions express lymphatic markers LYVE-1, PROX1, podoplanin and VEGFR-3. The smooth muscle–like cells of AMLs are morphologically and immunohistochemically similar to LAM cells, including reactivity with antibodies directed against actin, desmin, vimentin, and HMB-45 as well as estrogen and progesterone receptors. Unlike the dilated airspaces in emphysema, the cystic spaces found in LAM may be partially lined with hyperplastic type II cells.
Chest radiograph
The chest radiograph may appear relatively normal, even late in the disease, or may suggest hyperinflation only. As the disease progresses, the chest radiograph often demonstrates diffuse, bilateral and symmetric reticulonodular opacities, cysts, bullae or a "honeycomb" (i.e., pseudo fibrotic) appearance. Pleural effusion and pneumothorax may be apparent. Preservation of lung volumes in the presence of increased interstitial markings is a radiographic hallmark of LAM that helps distinguish it from most other interstitial lung diseases, in which alveolar septal and interstitial expansion tend to increase the lung’s elastic recoil properties and decreased lung volumes.
High-resolution computed tomography
The high-resolution computed tomography (HRCT) chest scan is better than the chest radiograph to detect cystic parenchymal disease and is almost always abnormal at the time of diagnosis, even when the chest radiograph and pulmonary function assessments are normal. The typical CT shows diffuse round, bilateral, thin-walled cysts of varying sizes ranging from 1 to 45 mm in diameter. The numbers of cysts varies in LAM from a few to almost complete replacement of normal lung tissue. The profusion of cysts tends to be milder in patients with TSC-LAM than S-LAM, perhaps explained in part because TSC-LAM patients typically receive earlier screening. Pleural effusions are seen on CT in 12% of patients with S-LAM and 6% of patients with TSC-LAM. Other CT features include linear densities (29%), hilar or mediastinal lymphadenopathy (9%), pneumothorax, lymphangiomyoma, and thoracic duct dilation. Ground-glass opacities (12%) suggest the presence of interstitial edema due to lymphatic congestion. In patients with TSC, nodular densities on HRCT may represent multifocal micronodular pneumocyte hyperplasia (MMPH) made up of clusters of hyperplastic type II pneumocytes. MMPH may be present in males or females with TSC in the presence or absence of LAM, but not in patients with S-LAM. MMPH is not typically associated with physiologic or prognostic consequences, but one case of respiratory failure due to MMPH has been reported.
Ventilation-perfusion scans
In one study ventilation-perfusion scans were abnormal in 34 of 35 LAM patients. The most common abnormality was nonspecific diffuse heterogeneity, usually grossly matched. These authors also described an “unusual,” “speckling pattern” on the perfusion images in 74% of patients, consisting of “small, often peripheral collections of radioisotope.”
Positron emission tomography
LAM and AML lesions do not typically exhibit increased uptake of 18F-fluorodeoxyglucose on positron emission tomography (PET) scanning. Other neoplasms (or sources of inflammation) should therefore be considered in known or suspected LAM cases in which FDG-PET results are positive.
Abdominal imaging
Abnormalities on abdominal imaging, such as renal AML and enlarged lymphatic structures, are also common in LAM. Fat density within a renal mass is pathognomonic of AMLs. AMLs are more prevalent and more frequently bilateral and large in patients with TSC-LAM than in patients with S-LAM. AML size correlates with the prevalence of pulmonary cysts in patients with TSC. One study CT imaged 256 patients with S-LAM and 67 with TSC-LAM. Renal AMLs were present in 32% of patients with S-LAM and 93% of patients with TSC-LAM. Hepatic AMLs were present in 2% of patients with S-LAM and 33% of patients with TSC-LAM. Ascites was uncommon, seen in fewer than 10% of patients with LAM. Abdominal lymphangiomatosis, often containing both cystic and solid components, were seen in 29% of patients with S-LAM and 9% of patients with TSC-LAM.
Central nervous system imaging
Central nervous system abnormalities, such as cortical or subependymal tubers and astrocytomas, are common in patients with TSC, including those with TSC-LAM, but are not found in women with S-LAM. Moss and associates reported that women with S-LAM and TSC-LAM may have an increased incidence of meningioma, but the significance of that finding has been challenged.
Pulmonary function studies
Pulmonary function testing in patients with LAM may be normal or may reveal obstructive, restrictive or mixed patterns. Obstructive physiology is the most common abnormality. Quality-controlled lung function data were collected prospectively by the NHLBI Registry, a 5-year study of patients with LAM in centers around the United States. Spirometry revealed obstructive changes in about 57% of patients and normal results in 34%. Restriction, defined as a total lung capacity less than the lower limit of normal, was seen in 11%. Hyperinflation was present in about 6%. The average residual volume was 125% of predicted when measured by plethysmography, but was only 103% of predicted determined with gas dilution methods, suggesting significant air trapping in noncommunicating airspaces. Approximately 25% of patients with obstructive physiology may demonstrate bronchodilator responsiveness but may be less in more severe obstruction. The obstructive physiologic defect in LAM is primarily attributable to airflow obstruction. The earliest change in initial pulmonary function testing in various case series was abnormal gas transfer, as assessed by the diffusing capacity for carbon monoxide (DLCO), described in 82% to 97% of patients. It is not unusual for DLCO to be reduced out of proportion to forced expiratory volume in 1 second (FEV1). Reduction in DLCO and increase in residual volume are generally considered to be LAM's earliest physiologic manifestations.
Cardiopulmonary exercise testing in a much larger cohort of patients with LAM revealed a reduced maximal oxygen consumption (VO2 max) and anaerobic threshold in 217 patients. Exercise-induced hypoxemia was found even in patients who did not have resting abnormalities in FEV1 and DLCO. In most patients, exercise was thought to be ventilation limited, owing to airflow obstruction and increased dead-space ventilation.
Disease progression is usually accompanied by a progressive obstructive ventilatory defect. Decline in FEV1 is the most commonly used parameter to monitor disease progression. Although resting pulmonary hypertension appears to be unusual in LAM, pulmonary arterial pressure often rises with low levels of exercise, related in part to hypoxemia. One study reported an increase in intraparenchymal shunts in dyspneic patients with LAM, which may contribute to resting and exercise hypoxemia.
Diagnosis
LAM can come to medical attention in several ways, most of which trigger a chest CT. Thin-walled cystic change in the lungs may be found incidentally on CT scans of the heart, chest or abdomen (on the cuts that include lung bases) obtained for other purposes. HRCTs of TSC patients reveals that about 20% of women have cystic change by age 20 and about 80% of women have cystic changes after age 40. LAM is sometimes revealed by chest CT in patients who present with an apparent primary spontaneous pneumothorax, but more often CT scanning is not ordered (in the United States) until recurrences occur. Progressive dyspnea on exertion without the exacerbations and remissions that are characteristic of asthma or COPD sometimes prompt a chest CT. A review of the CT by an expert familiar with LAM may increase diagnostic accuracy. Chylothorax can also bring LAM to attention.
In some cases, a LAM diagnosis can be made with confidence on clinical grounds (without biopsy) in patients with typical cystic changes on high resolution CT scanning of the lung and findings of tuberous sclerosis, angiomyolipoma, lymphangioleiomyoma, chylothorax or serum VEGF-D > 800 pg/ml.
If none of these clinical features are present, a biopsy may be necessary to make the diagnosis. Video-assisted thoracoscopic lung biopsy is the most definitive technique, but transbronchial biopsy has a yield of over 50% and can also be effective. The safety of the latter procedure in patients with diffuse cystic disease and the profusion of cystic change that predicts an informative biopsy are incompletely understood, however. Cytology of chylous fluids, aspirated abdominal nodes or lymphatic masses can also be diagnostic.
Diagram 1 outlines a proposed algorithm for the diagnosis of LAM.
Pneumothorax
Pneumothoraces in LAM patients tend to recur, especially after conservative management such as observation, aspiration or simple tube thoracostomy. Over 65% of LAM patients develop pneumothorax during the course of their illness, averaging 3.5 pneumothoraces in those who have at least one pneumothorax. The LAM Foundation Pleural Consensus Group advocated the use of a pleural symphysis procedure with the first pneumothorax, given the greater than 70% chance of recurrence. Chemical sclerosis, mechanical abrasion, talc poudrage and pleurectomy have been effective in patients with LAM, but mechanical abrasion is preferred for those who may require pulmonary transplantation in the future. About half of LAM patients who have undergone transplant have had a prior pleurodesis procedure, and more than 75% of those had had prior bilateral pleurodesis. Although pleurodesis is not a contraindication to transplantation, it can result in increased perioperative bleeding.
Chylothorax
Chyle does not generally cause pleural inflammation or fibrosis. Small stable chylous effusions rarely require intervention once the LAM diagnosis is made. Shortness of breath may mandate possibly repeated drainage. Sirolimus is effective for chylous effusions and most experts believe it should be used as the first line of therapy. Imaging the source of the leak with heavy T2-weighted MRI or contrast lymphangiography is an advised for refractory effusions. Some leaks are amenable to embolization through catheters threaded from groin lymph nodes into the thoracic duct. Thoracic duct ligation can be considered, but since thoracic effusions sometimes originate from ascites that are siphoned into the chest by the bellows action of the thorax, it is important to rule out an abdominal source before considering this option. Pleural symphysis may be required to prevent nutritional and lymphocyte deficiencies that can result from repeated taps or persistent drainage. Chemical pleurodesis is generally an effective therapy for chylothorax, as is mechanical abrasion and talc poudrage.
Angiomyolipoma
Renal angiomyolipomas (AMLs) may require embolization or cauterization for control of bleeding, a complication that is thought to be more common when tumor diameter exceeds 4 cm. The extent of aneurysmal change may determine bleeding risk. Serial abdominal imaging should be performed to assess AML size at 6- to 12-month intervals, at least until trends in growth are clear. Nephron sparing partial resections may be considered for very large tumors. Nephrectomy is sometimes required for tumors with intravascular extension or other reasons, but is rarely the approach of choice for AMLs that can be managed by less invasive means. Everolimus is approved by the U.S. Food and Drug Administration (FDA) for AML treatment.
Lymphangioleiomyoma
Lymphangioleiomyomatoses are fluid-filled hypodense structures present in the retroperitoneal regions of the abdomen and pelvis in about 30% of LAM patients. They generally do not require intervention. Biopsy or resection can lead to prolonged leakage. mTOR inhibitors are effective at shrinking the size of lymphangioleiomyomatosis, and can lead to total resolution.
Management-other
Estrogen-containing medications can exacerbate LAM and are contraindicated. Agents that antagonize the effects of estrogen have not been proven to be effective for treatment, but no proper trials have been done. A trial of bronchodilators should be considered in LAM patients, because up to 17% to 25% have bronchodilator-responsive airflow obstruction. Oxygen should be administered to maintain oxyhemoglobin saturations of greater than 90% with rest, exercise and sleep. Bone densitometry should be considered in all patients who are immobilized and/or on antiestrogen therapies, and appropriate therapy instituted for osteoporotic patients. Proper attention should be paid to cardiovascular health following natural or induced menopause. Immunizations for pneumococcus and influenza should be kept up to date. Pulmonary rehabilitation seems to be particularly rewarding in young, motivated patients with obstructive lung disease, but studies to assess this intervention's effect on exercise tolerance, conditioning and quality of life have not been done.
Pharmacological treatment
Sirolimus is an mTOR inhibitor that stabilizes lung function and improves some measures of life in LAM patients. It is approved by the FDA for use in LAM, based on the results of the Multicenter International LAM Efficacy and Safety of Sirolimus (MILES) Trial. MILES data supports the use of sirolimus in patients who have abnormal lung function (i.e. FEV1<70% predicted). Whether the benefits of treatment outweigh the risks for asymptomatic LAM patients with normal lung function is not clear, but some physicians consider treatment for declining patients who are approaching the abnormal range for FEV1. Sirolimus also appears to be effective for the treatment chylous effusions and lymphangioleiomyomatosis. The benefits of sirolimus only persist while treatment continues. The safety of long term therapy has not been studied.
Potential side effects from mTOR inhibitors include swelling in the ankles, acne, oral ulcers, dyspepsia, diarrhea, elevation of cholesterol and triglycerides, hypertension and headache. Sirolimus pneumonitis and latent malignancy are more serious concerns, but occur infrequently. Sirolimus inhibits wound healing. It is important to stop therapy with the drug for 1–2 weeks before and after elective procedures that require optimal wound healing. Precautions must be taken to avoid prolonged sun exposure due to increased skin cancer risk.
Treatment with another mTOR inhibitor, everolimus, was reported in a small, open-label trial to be associated with improvement in FEV1 and six-minute walk distance. Serum levels of VEGF-D and collagen IV were reduced by treatment. Adverse events were generally consistent with those known to be associated with mTOR inhibitors, although some were serious and included peripheral edema, pneumonia, cardiac failure and Pneumocystis jirovecii infection. Escalating doses of everolimus were used, up to 10 mg per day; higher than what is typically used clinically for LAM.
Serum VEGF-D concentration is useful, predictive and prognostic biomarker. Higher baseline VEGF-D levels predicts more rapid disease progression and a more robust treatment response.
Hormonal approaches to treatment have never been tested in proper trials. In the absence of proven benefit, therapy with progesterone, GnRh agonists (e.g., Lupron, goserelin) and tamoxifen are not routinely recommended. Doxycycline had no effect on the rate of lung function decline in a double blind trial.
Sirolimus is often effective as first-line management for chylothorax. If chylous leakage or accumulations persist despite treatment, imaging with heavy T2 weighted MRI, MRI lymphangiography or thoracic duct lymphangiography can be considered. Pleural fusion procedures can be considered in refractory cases.
Pregnancy
Pregnancy has been reported to exacerbate LAM in some cases. However, the risk has not been rigorously studied. In a survey of 318 patients who indicated that they had had at least one pregnancy, 163 responded to a second survey focusing on lung collapse. A total of 38 patients reported a pneumothorax with pregnancy, consistent with an incidence of pneumothorax in pregnancy of at least 10% (38 of 318). In one third of patients, the pneumothorax during pregnancy led to the LAM diagnosis. Pneumothoraces were almost twice as frequent on the right as on the left, and four women presented with bilateral spontaneous pneumothorax. Most pneumothoraces took place during the second and third trimesters. This study and others suggest that pregnancy is associated with pleural complications in LAM patients. Few women with a known LAM diagnosis choose to become pregnant and patients in whom LAM is diagnosed during pregnancy rarely have baseline pulmonary function tests available, complicating resolution of this question.
Air travel
Air travel is not typically restricted in LAM patients. In a 276 patient survey, eight cases of radiographically documented pneumothorax were associated with 454 flights. In five of the eight, symptoms may have been present before boarding. Other symptoms and signs, including anxiety (22%), chest pain (12%), shortness of breath (14%), cyanosis (2%) and hemoptysis (0.4%), were noted in 10% to 20% of flights. The study concluded that air travel is well tolerated by most patients.
A study of 281 patients with LAM who had routine chest radiography identified seven with acute pneumothorax. No difference emerged in the incidence of pneumothorax in patients who traveled by ground versus air. In advising patients, it is reasonable to consider history of frequent or recent pneumothoraces and the overall extent of cardiopulmonary impairment. Patients with poor cardiopulmonary reserve may tolerate even small pneumothoraces poorly. It is prudent to seek medical evaluation, including a chest radiograph, before flying if pleuritic chest pain or unexplained shortness of breath is present. Hypoxemia during flight presents independent risks. Patients should consult with their physicians regarding on-board oxygen use. In most cases, however, air travel in patients is not restricted.
Prognosis
Survival estimates vary, dependent on mode of presentation or ascertainment, and have generally trended upward, probably due to earlier recognition through more widespread use of CT scanning. In a recent population-based cohort survey, median survival was found to be 29 years. Data from earlier, large case series indicated that 38% to 78% of patients were alive at 8.5 years from the time of disease onset.
Patients typically develop progressive airflow obstruction. In a cohort of patients in the United Kingdom, 10 years after symptom onset, 55% of 77 patients were breathless walking on flat ground and 10% were housebound. The average annual rate of decline in FEV1 and DLCO in 275 patients studied in a single pulmonary function laboratory at the NHLBI was 75 ± 9 mL, and 0.69 ± 0.07 mL/min/mm Hg, respectively. In other series from Europe, the rate of decline in FEV1 was considerably higher, estimated at approximately 100 to 120 mL/yr. In the MILES trial, patients in the placebo group lost 134 cc/yr. There was some evidence in these studies that rate of decline in lung function correlates with initial DLCO, with menopausal status and high baseline VEGF-D.
Epidemiology
LAM is almost completely restricted to women. While lung cysts consistent with LAM are reported in some men with tuberous sclerosis, very few of these men develop symptoms. The prevalence of LAM is estimated using data from registries and patient groups and is between 3.4-7.8/million women. The number of new cases each year is between 0.23-0.31/million women/year in the US, UK and Switzerland. The variation between countries and between adjacent states in the US, suggest that a significant number of women with LAM remain either undiagnosed or their symptoms are attributed to other diseases. Adult women with tuberous sclerosis are more likely to develop LAM than women without tuberous sclerosis. Cohorts of patients with tuberous sclerosis have been screened for LAM using CT scanning. In a retrospective study of adults with tuberous sclerosis, CT demonstrated lung cysts in 42% of 95 women and 13% of 91 men. In general, lung cysts were larger and more numerous in women than in men. In a further retrospective study of women with TSC who underwent CT scanning to detect LAM, 25% of those in their 20s had lung cysts whereas 80% of women in their 40s were affected, suggesting that the development of LAM is age dependent at least in tuberous sclerosis-related LAM. Although the prevalence of tuberous sclerosis at 1 in 6000 births is much greater than that of LAM, most pulmonary clinics see more cases of sporadic than tuberous sclerosis-LAM: probably due to a combination of low levels of screening for LAM in tuberous sclerosis and in many, the absence of symptoms.
Female sex and tuberous sclerosis are the only known risk factors. Although use of supplemental estrogen is not associated with development of LAM, one study suggested that use of estrogen-containing contraceptive pills was associated with earlier onset.
Clinical research
The Rare Lung Diseases Consortium (RLDC) studies LAM. The RLDC is part of the Rare Diseases Clinical Research Network (RDCRN), an initiative of the Office of Rare Diseases Research (ORDR) of the National Center for Advancing Translational Sciences (NCATS), one of the centers of the US National Institutes of Health (NIH).
Patient registry
LAM patients, families, and caregivers are encouraged to join the NIH Rare Lung Diseases Consortium Contact Registry. This is a privacy-protected site that provides information for individuals interested in the latest scientific news, trials and treatments related to rare lung diseases. They can register with the LAM Foundation to track the LAM community and to connect with other patients and families.
In popular culture
In "Lucky Thirteen", the fifth episode of the fifth season of House, Spencer (Angela Gots) was diagnosed with LAM, though later it was found to be a case of Sjögren's syndrome.