
 | ||
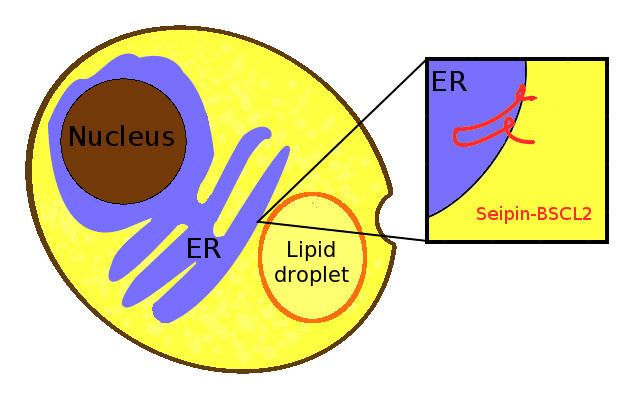
Seipin is a homo-oligomeric integral membrane protein in the endoplasmic reticulum (ER) that concentrates at junctions with cytoplasmic lipid droplets (LDs). Alternatively, seipin can be referred to as Bernardinelli-Seip congenital lipodystrophy type 2 protein (BSCL2), and it is encoded by the corresponding gene of the same name, i.e. BSCL2. At protein level, seipin is expressed in cortical neurons in the frontal lobes, as well as motor neurons in the spinal cord. It is highly expressed in areas like the brain, testis and adipose tissue. Seipin's function is still unclear but it has been localized close to lipid droplets, and cells knocked out in seipin which have anomalous droplets. Hence, recent evidence suggests seipin to play a crucial role in lipid droplet biogenesis.
Contents
Function
Initially dubbed "mysterious protein", recent empirical studies gradually start to unveil some of seipin's most compelling physiological functions. Among these, the following have been identified: central regulation of energy homeostasis, lipid catabolism (essential for adipocyte differentiation), lipid storage and lipid droplet maintenance, as well as prevention of ectopic lipid droplet formation in non-adipose tissues. Additionally, mutations of BSCL2 have been recently linked to the Silver-Syndrome and Celia's Encephalopathy.
Primary structure
The seipin gene BSCL2 was originally identified in mammals and the fruit fly, and later extended to fungi and plants. The human seipin gene is located on chromosome 11q13, with protein coding on the Crick strand.
There are three validated coding transcripts in GenBank. The primary transcript originally described, contained 11 exons with protein coding beginning on exon 2 and ending in exon 11 (transcript variant 2), resulting in a 398 amino acid protein with two strongly predicted transmembrane domains (TMDs), coded in exons 2 and 7 (isoform 2).
However, a longer transcript (variant 1) is generated with an alternative first exon containing a translational start site that results in an additional 64 amino acids at the N-terminal extension, 462 amino acids in total (isoform 1).
A third coding transcript (variant 3) splices out exon 7 and produces a shortened and altered carboxy terminus in exon 10, generating a protein of 287 amino acids (isoform 3).
Secondary structure
The secondary structure of seipin includes a conserved central core domain, and diverse cytosolic N- and C-termini.
Tertiary structure
Seipine’s tridimensional structure has 5 different domains:
·Topological domain: located at the cytoplasmic region, includes the first 26 amino acids.
MVNDPPVPAL LWAQEVGQVL AGRARR·Transmembrane domain: chain of 21 aminoacids (27-47) with helical conformation.
LLLQFGVLFC TILLLLWVSVF·Topological domain: located at the lumenal region, it covers the longest region of amino acids -195 (positions 48-242).
LYGSFYYSYMPTV SHLSPVHFYY RTDCDSSTTS LCSFPVANVS LTKGGRDRVLMYGQPYRVTL ELELPESPVN QDLGMFLVTI SCYTRGGRII STSSRSVMLHYRSDLLQMLD TLVFSSLLLF GFAEQKQLLE VELYADYREN SYVPTTGAIIEIHSKRIQLY GAYLRIHAHF TGLRYLLYNF PMTCAFIGVA SN·Transmembrane domain: as the previous transmembrane domain, and with the same length of amino acid, the protein located between the two regions of the cell adopts an helical conformation (positions 243-263).
FTFLSVIVLF SYMQWVWGGIW·Topological domain: last sequence of amino acids (264-398) located at the cytoplasmic side.
PRHRFSLQVN IRKRDNSRKE VQRRISAHQP GPEGQEESTP QSDVTEGESP EDPSGTEGQL SEEEKPDQQP LSGEEELEPE ASDGSGSWE DAALLTEANL PAPAPASASAP VLETLGSSEP AGGALRQRPT CSSSPathophysiology
There are three different variations of seipin's amino acid sequence:
All seipin mutations occur within its loop domain. Between some of these, four large deletions can be found which indicate that at least exons 4 and 5 are required for seipin function in humans. In addition, other six mutations have been identified in the loop domain. The majority of these cluster at the single asparagine-linked glycosylation site (NVS) in seipin. The two mutations that cause neuronal seipinopathy, N88S and S90L, are located directly within this site. Apart from suspending the glycosylation process, these mutations engender an aggregation of seipin and, consequently, the initiation of the ER stress response. The seipin protein can also have a modification residue, that can transform the 289’ and 372’ serine into a phosphoserine, an ester of serine and phosphoric acid.
Overexpression of mutated seipin proteins N88S or S90L can also activate autophagy, and substantially altering the sub-cellular distribution of the autophagosome marker GFP-LC3, which leads to a number of large vacuoles appearing in the cytoplasm. The sub-cellular location of GFP-LC3 and mutated seipin proteins highly overlap. Moreover, these seipin proteins can diffuse small lipid droplets to fuse into larger lipid.
Seipin mutations have been associated with congenital generalized lipodystrophy (see below), and mutations in an N-glycosylation motif links seipin to two other disorders, i.e. Silver syndrome and autosomal-dominant distal hereditary motor neuropathy type V.
Congenital generalized lipodystrophy
CGL (congenital generalized lipodystrophy) is a heterogeneous genetic disorder characterized by almost complete loss of adipose tissue (both metabolic and mechanical adipose depots) and an increase of ectopic fat storage in liver and muscle. Of the four CGL types, BSCL2 (Berardinelli-Seip Congenital lipodystrophy type 2), resulting from mutations in the BSCL2/seipin gene, exhibits the most severe lipodystrophic phenotype.
Furthermore, these patients could suffer dyslipidemia, hepatic steatosis, insulin resistance and hypertrophic cardiomyopathy due to a cell-autonomous defect in cardiomyocytes.
Neurological seipinopathies
For many years mutations of the seipin gene were associated with a loss of function, such as in CGL (see above). However, recent studies show that mutations such as N88S and S90L seem to have a gain-of-toxic-function which may result in autosomal dominant motor neuron diseases and distal hereditary motor neuropathy.
Owing to the wide clinical spectrum of these mutations, it has been proposed to collectively refer to seipin-related motor neuron diseases as seipinopathies.
Symptoms can vary and include: developmental regression of motor and cognitive skills in the first years of life leading to death (encephalopathy), muscle weakness and spasticity in lower limbs (spastic paraplegia type XVII), weakness of distal muscles of upper limbs (distal hereditary motor neuropathy type V) as well as wasting of the hand muscles (in both cases). Complex forms of seipinopathies may include deafness, dementia or mental retardation.
Male infertility
Testicular tissue-derived seipin is essential for male fertility by modulating testicular phospholipid homeostasis. The lack of seipin in germ cells results in complete male infertility and teratozoospermia. Spermatids devoid of seipin in germ cells are morphologically abnormal with large ectopic lipid droplets and aggregate in dysfunctional clusters. Elevated levels of phosphatidic acid accompanied with an altered ratio of polyunsaturated to monounsaturated and saturated fatty acids show impaired phospholipid homeostasis during spermiogenesis .