
Abbreviations Palmitamide MEA Formula C18H37NO2 Boiling point 461.5 °C | Related compounds Density 910 kg/m³ Appearance White crystals | |
 | ||
Pharmacology of palmitoylethanolamide
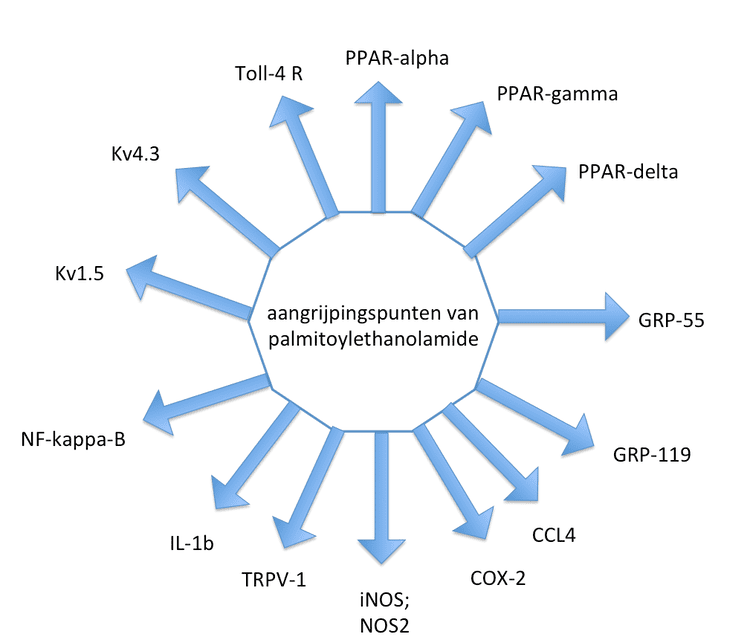
Palmitoylethanolamide (PEA) is an endogenous fatty acid amide, belonging to the class of nuclear factor agonists. PEA has been demonstrated to bind to a receptor in the cell-nucleus (a nuclear receptor) and exerts a great variety of biological functions related to chronic pain and inflammation. The main target is thought to be the peroxisome proliferator-activated receptor alpha (PPAR-α). PEA also has affinity to cannabinoid-like G-coupled receptors GPR55 and GPR119. PEA cannot strictly be considered a classic endocannabinoid because it lacks affinity for the cannabinoid receptors CB1 and CB2. However, the presence of PEA (and other structurally related N-acylethanolamines) has been known to enhance anandamide activity by a so-called "entourage effect".
Contents
- Pharmacology of palmitoylethanolamide
- Palmitoylethanolamide in nerve compression syndromes video abstract 93106
- Early studies
- Animal models
- Animal models of chronic pain and inflammation
- PEAs mechanism on non neuronal cells
- Clinical relevance
- Metabolism
- References
Several papers have demonstrated that an imbalance of the endocannabinoid system (ECS) and alterations in the levels of PEA occur in acute and chronic inflammation. For instance during β-amyloid-induced neuroinflammation the deregulation of cannabinoid receptors and its endogenous ligands accompanies the development and progression of disease.
PEA has been shown to have anti-inflammatory, anti-nociceptive, neuroprotective, and anticonvulsant properties.
Palmitoylethanolamide in nerve compression syndromes video abstract 93106
Early studies
Indications as anti-inflammation and analgesia stem from before 1980, and the birth of the molecule was in 1957. In that year 5 researchers from MSD described N-(2-hydroxyethyl)-palmitamide, as they called the molecule at that time, as a natural anti-inflammatory agent. They stated: " We have succeeded in isolating a crystalline anti-inflammatory factor from soybean lecithin and identifying it as (S)-(2-hydroxyethyl)-palmitamide. The compound also was isolated from a phospholipid fraction of egg yolk and from hexane-extracted peanut meal."
In 1975 Czech physicians described the result of a clinical trial in joint pain in The Lancet. The analgesic action of 3 grams of aspirin during the day was compared to PEA 1.8 gram/day. Both drugs were reported to enhance joint movements and decrease pain. In 1970 the pharmaceutical industry Spofa introduced Impulsin, tablets of 500 mg PEA for the treatment and prophylaxis of flu and respiratory infections in Czechoslovakia, and the company Almirall introduced Palmidrol, as tablets and as a suspension in Spain in 1976 for the same indication.
In the 1990s, the relation between anandamide and PEA was described, and the expression of receptors sensitive for those two molecules on mast cells was first demonstrated by the group of Nobel prize winner Rita Levi-Montalcini. In this period more insight into the function of the endogenous fatty acid derivatives emerged, and compounds such as oleamide, palmitoylethanolamide, 2-lineoylglycerol, 2-palmitoylglycerol were explored for their capacity to modulate pain sensitivity and inflammation via what at that time was thought to be, the endocannabinoid signalling pathway. One group demonstrated that PEA could alleviate, in a dose-dependent manner, pain behaviors elicited in mice-pain models and could downregulate hyperactive mast cells. PEA and related compounds such as anandamide also seem to have synergistic effects in models of pain and analgesia.
Animal models
In a variety of animal models PEA seems promising, and researchers could demonstrate relevant clinical activity in a variety of disorders, from multiple sclerosis to neuropathic pain.
In the mouse forced swimming test palmitoylethanolamide was comparable to fluoxetine in anti-depressant effects. An Italian study published in 2011 found that PEA reduced the raised intraocular pressure in glaucoma. In a spinal trauma model, PEA could reduce neurological deficit through the reduction of mast cell infiltration and activation. PEA in this model also reduced the activation of microglia and astrocytes. Its activity as an inhibitor of inflammation could counteracts reactive astrogliosis induced by beta-amyloid peptide, in a model relevant for neurodegeneration, probably via the PPAR-α mechanism of action. In models of stroke and other traumata of the central nervous system, PEA exerted neuroprotective properties. For pet animals PEA has been used successfully to treat painstates and chronic inflammation.
Animal models of chronic pain and inflammation
Chronic pain and neuropathic pain are indications for which there is high unmet need in the clinic. PEA has been tested in a variety of animal models for chronic and neuropathic pain. As cannabinoids, such as THC, have been proven to be effective in neuropathic pain states. The analgesic and antihyperalgesic effects of PEA in two models of acute and persistent pain seemed to be explained at least partly via the de novo neurosteroid synthesis. In chronic granulomatous pain and inflammation model, PEA could prevent nerve formation and sprouting, mechanical allodynia, and PEA inhibited dorsal root ganglia activation, which is a hallmark for winding up in neuropathic pain. The mechanism of action of PEA as an analgesic and anti-inflammatory molecule is probably based on different aspects. PEA inhibits the release of both preformed and newly synthesised mast cell mediators, such as histamine and TNF-alpha. PEA, as well as its analogue adelmidrol (di-amide derivative of azelaic acid), can both down-regulate mast cells. PEA reduces the expression of cyclooxygenase-2 (COX-2) and inducible nitric oxide synthase (iNOS) and prevents IkB-alpha degradation and p65 NF-kappaB nuclear translocation, the latter related to PEA as an endogenous PPAR-alpha agonist. In 2012 it became clear that PEA can also reduce reperfusion injury and the negative impact of shock on various outcome parameters, such as renal dysfunction, ischemic injury and inflammation, most probably via the PPAR-alpha pathway. Among the reperfusion and inflammation markers measured PEA could reduce the increase in creatinine, γGT, AST, nuclear translocation of NF-κBp65; kidney MPO activity and MDA levels, nitrotyrosine, PAR and adhesion molecules expression, the infiltration and activation of mastcells and apoptosis.
PEA seems to be produced in human as well as in animals as a biological response and a repair mechanism in chronic inflammation and chronic pain. In a model of visceral pain (inflammation of the urinary bladder) PEA was able to attenuate the viscero-visceral hyper-reflexia induced by inflammation of the urinary bladder, one of the reasons why PEA is currently explored in the painful bladdersyndrome. In a different model for bladder pain, the turpentine-induced urinary bladder inflammation in the rat, PEA also attenuated a referred hyperalgesia in a dose-dependent way. Chronic pelvic pain in patients seem to respond favourably to a treatment with PEA.
PEA's mechanism on non-neuronal cells
PEA, as an N-acylethanolamine, has physico-chemical properties comparable to anandamide, and while it is not strictly an endocannabinoid, it is often studied in conjunction with anandamide because of their overlapping synthetic and metabolic pathways. N-acylethanolamines like PEA often act as signaling molecules, activating intracellular and membrane-associated receptors to regulate a variety of physiological functions. The signaling lipid PEA is known to activate intracellular, nuclear and membrane-associated receptors and regulate many physiological functions related to the inflammatory cascade and chronic pain states. Endocannabinoid lipids like PEA are widely distributed in nature, in a variety of plant, invertebrate, and mammalian tissues.
PEA's mechanism of action sometimes is described as Autacoid Local Injury Antagonism (acronym ALIA), and PEA under this nomenclature is an ALIAmide. It was the group of the Nobel prize laureate Rita Levi-Montalcini who in 1993 first presented evidence supporting that lipid amides of the N-acylethanolamine type (such as PEA) are potential prototypes of naturally occurring molecules capable of modulating mast cell activation, and her group coined the acronym ALIA in that paper. An autocoid is a regulating molecule, locally produced. An ALIAmide is an autocoid synthesized on-demand in response to injury, and acts locally to counteract such pathology. The mast cell soon after the breakthrough paper of Levi-Montalcini appeared to be an important target for the anti-inflammatory activity of PEA, and since 1993, at least 25 papers have been published on the various effects of PEA on the mast cell. Mast cells are often found in proximity to sensory nerve endings and their degranulation can enhance the nociceptive signal, the reason why peripheral mast cells are considered to be pro-inflammatory and pro-nociceptive. PEA's activity is currently seen as a new inroad in the treatment of neuropathic pain and related disorders based on overactivation of glia and glia-related cells, such as in diabetes and glaucoma. Microglia plays a key role in the winding up phenomena and central sensitization.
Clinical relevance
PEA has been explored in humans; spanning various clinical trials in a variety of pain states, for inflammatory and pain syndromes. Its positive influence in atopic eczema for instance seems to originate from PPAR alpha activation. PEA is available for human use as a supplement (400 mg capsules) and as food for medical purposes In Italy and Spain (300 mg and 600 mg tablets).
In a 2012 review, all clinical trials to date were summarized. In a 2015 analysis of a double blind placebo controlled study of PEA in sciatic pain, the Numbers Needed to Treat was 1.5. Its positive influence in chronic pain, and inflammatory states such as atopic eczema, seems to originate mainly from PPAR alpha activation. Since 2012 a number of new trials have been published, among which studies in glaucoma. PEA also seems to be one of the factors responsible for the decrease in pain sensitivity during and after sport, comparable to the endogenous opiates (endorphines).
From a clinical perspective the most important and promising indications for PEA are linked to neuropathic and chronic pain states, such as diabetic neuropathic pain, sciatic pain, CRPS, pelvic pain and entrapment neuropathic painstates. In a blind pilot trial in 25 patients affected by temporomandibular joint's (TMJ) osteoarthritis or synovitis pain, patients were randomly to PEA or ibuprofen 600 mg three times a day for two weeks. Pain decrease after two weeks of treatment was significantly higher in PEA treated patients than in patients receiving the NSAID (p=0.0001) Masticatory function also improves more on PEA compared to the NSAID. In 2012, 20 patients suffering from thalidomide and bortezomib induced neuropathy were reported to have improved nerve functions and less pain after a two months treatment with PEA 600 mg daily. The authors pointed out that although a placebo effect might play a role in the reported pain relief, the changes in neurophysiological measures clearly indicated that PEA exerted a positive action on the myelinated fibre groups. 30 patients suffering from neuropathic pain, which were refractory to treatment with analgesics, included pregabalin, were responding well in 45 days, with a decrease of painscores > 50% when pregabalin was tapered in again, up to 600 mg/day in combination with PEA, without signs of drug-drug interaction. In 2013 a review was published on the clinical efficacy and safety of PEA in flu, based on a 6 double blind placebo controlled RCT's. This review underscores the anti-inflammatory action of PEA. These effects are also the explanation of PEA's retinoprotectant effects. PEA is available as capsules (400 mg), tablets (300 and 600 mg) and as cream (0,3% and 1.5%).
Metabolism
PEA is metabolized by cellular enzymes, fatty acid amide hydrolase (FAAH) and N-acylethanolamine-hydrolyzing acid amidase (NAAA), the latter of which has more specificity toward PEA over other fatty acid amides. To date, no drug interactions have been reported in the literature, nor any clinical relevant or dose-limiting side effect.