
Formula [C21H28N6O18P3]+ | Molar mass 745.398 g/mol | |
 | ||
Nicotinic acid adenine dinucleotide phosphate, (NAADP), is a Ca2+-mobilizing second messenger synthesised in response to extracellular stimuli. Like its mechanistic cousins, IP3 and cyclic adenosine diphosphoribose (Cyclic ADP-ribose), NAADP binds to and opens Ca2+ channels on intracellular organelles, thereby increasing the intracellular Ca2+ concentration which, in turn, modulates sundry cellular processes (see Calcium signalling). Structurally, it is a dinucleotide that only differs from the house-keeping enzyme cofactor, NADP by a hydroxyl group (replacing the nicotinamide amino group) and yet this minor modification converts it into the most potent Ca2+-mobilizing second messenger yet described. NAADP acts across phyla from plants to man.
Contents
Discovery
In their landmark 1987 paper, Hon Cheung Lee and colleagues discovered not one but two Ca2+-mobilizing second messengers, cADPR and NAADP from the effects of nucleotides on Ca2+ release in sea urchin egg homogenates. It turns out that NAADP was a contaminant in commercial sources of NADP, but it was not until 1995 that its structure was solved. The first demonstration that NAADP could act in mammalian cells (pancreas) came four years later. Subsequently, NAADP has been detected in sources as diverse as human sperm, red and white blood cells, liver, and pancreas, to name but a few.
Kinetics and transduction
The first demonstration that NAADP levels increase in response to an extracellular stimulus arose from studying sea urchin fertilization (NAADP changed in both the eggs and sperm upon contact). Subsequently, other cell types have followed suit, as exemplified by the pancreas (acinar and beta cells), T-cells, and smooth muscle. Levels increase very rapidly — and possibly precede the increase in the other messengers IP3 and cADPR— but can be very transient (spiking and returning to basal levels within seconds). The transduction mechanisms that couple cell stimuli to such NAADP increases are ill-defined, with some suggestions of cyclic AMP or cytosolic Ca2+ itself stimulating synthesis.
Synthetic enzymes
Regardless of the details, an outstanding issue is that the physiological route of NAADP synthesis has still not been unequivocally identified — neither the reaction(s) nor the enzyme(s). Clearly, it is theoretically possible there may be multiple routes of synthesis, but this would be unprecedented in the second messenger world. To date, the most favoured hypothesis is the so-called base-exchange reaction (nicotinic acid + NADP → NAADP + nicotinamide; catalyzed by ADP-ribosyl cyclases) which are a family of enzymes that include CD38 and CD157 in mammals (and orthologs in sea urchin and Aplysia ovotestis). These were first discovered as the synthetic enzymes for cADPR but later revealed to be multifunctional, promiscuous enzymes that can also produce NAADP. Certainly NAADP production can occur in vitro but whether it occurs in vivo is another question (because genetic knockout or knock-down of ADP-ribosyl cyclases has no effect on NAADP production in some cell types), and there may be other routes which require different substrates and enzymes.
Degradative enzymes
Like any second messenger system, the signal must be terminated and there must be routes for NAADP removal but again, little is known with any degree of certainty. A 2'-3'-phosphatase stimulated by Ca2+ has been proposed in brain and, possibly in pancreatic acinar cells, that catabolises NAADP to inactive NAAD. More recently, CD38 has also been proposed to breakdown NAADP (to ADPRP — see inset). NAADP may also be reduced to NAADPH.
NAADP-selective physiology
Hardly surprisingly, the three major second messengers do not do the same thing and cannot always substitute for each other. The physiological consequences of Ca2+ release by each messenger may be different i.e. NAADP couples to downstream responses that cannot be mimicked by IP3 and cADPR. For example, NAADP selectively stimulates neuronal differentiation, or exocytosis in immune cells.
Target organelle
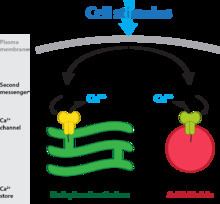
In contrast to IP3 and cyclic ADP-ribose which predominantly mobilize Ca2+ from the neutral and abundant endoplasmic reticulum (ER) store, NAADP selectively targets acidic Ca2+ stores — usually less abundant than the ER but with a pivotal role that belies their size. This paradigm shift away from the ER derives from seminal studies, again in sea urchin egg, that showed NAADP-mediated Ca2+ release was sensitive to agents that target acidic organelles (e.g. bafilomycin A1) but was less sensitive to ones that interfere with ER Ca2+ storage (e.g. thapsigargin).
Acidic Ca2+ store
This is a blanket term that encompasses a spectrum of acidic vesicles that include endosomes, lysosomes, and lysosome-related organelles and secretory vesicles and acidocalcisomes. They are a highly dynamic continuum of vesicles with a rich variety of established biochemical roles in cells, to which Ca2+ storage can now be added. Their luminal pH is one characteristic that distinguishes a given vesicle class from another: where endosomes are weakly acidic (pH 6-6.5), lysosomes are typically the most acidic (pH 4.5-5.0) and secretory vesicles are typically pH 5.5. Ca2+ is seen to be increasingly important for endo-lysosomal function, e.g. trafficking and autophagy. Aberrations in Ca2+ signals can have pathophysiological consequences, including lysosomal storage diseases such as Niemann Pick C and Mucolipidosis IV.
When NAADP mobilizes Ca2+ from these stores, the pH of the stores concomitantly increases (becomes more alkaline), as testified by studies in sea urchin egg, mammalian heart and pancreas. Whether this has consequences for vesicle (or NAADP) function remains to be seen, but luminal pH is usually crucial for resident protein activity.
Ca2+ uptake
In other Ca2+-storing organelles such as the endoplasmic reticulum or Golgi, stores are filled by calcium ATPase pumps, typified by the ubiquitous members of the SERCA or the SPCA (secretory pathway Ca2+-ATPase) families respectively. Ca2+ uptake by acidic stores occurs via other proteins: in yeast and plants (the best understood systems) the acidic vacuoles host two uptake pathways: a high affinity Ca2+-ATPase and a low affinity Ca2+/H+ antiporter (or exchanger, generically denoted as CHX). The pumps are different from the SERCA family (and, importantly, are insensitive to their inhibitor, thapsigargin) whereas the exchanger exploit the H+ gradient to drive Ca2+ uptake against its concentration gradient. The genes encoding these proteins are well-defined.
In higher organisms, the situation is less clear. Ca2+ uptake usually occurs via a thapsigargin-insensitive pathway (therefore precluding SERCA involvement) and appears to be dependent upon the H+ gradient; whether this occurs via a single (unknown) CHX or via exchangers in series (e.g. Na+/H+ exchanger coupled to a Na+/Ca2+ exchanger) is unproven. Acidic vesicles in some cell types may well take a leaf out of the yeasts'/plants' book and host two uptake pathways, but whether this is a widespread template is unclear.
In the absence of selective Ca2+ uptake inhibitors (often because we do not even know the protein/route), it is common to indirectly inhibit Ca2+ uptake by collapsing the thermodynamic drive (the H+ gradient). The H+ gradient can be eliminated either with H+ ionophores (protonophores) such as nigericin or monensin or by inhibiting the V-ATPase that generates the H+ gradient with compounds such as bafilomycin A1 or concanamycin.
Target channel (TPCs)
Even from the early pioneering work in sea urchin egg, it was clear from the pharmacological profile that NAADP acted upon a different channel from the IP3 receptor and ryanodine receptor and this has recently been borne out by the molecular identification of the NAADP receptor as members of the TPC (two-pore channel) family. As structural intermediates between single domain TRP and four-domain Voltage-dependent calcium channel, the TPCs form oligomers (possibly dimers) to form the functional Ca2+ channel. Appropriately, these channels reside on acidic organelles (including different classes of endosomes and lysosomes) likely due to the presence of endolysomal targeting sequences.
The effect of genetic manipulation of TPC levels (i.e. over-expression, knock-down or knock-out) is consistent with TPCs being the NAADP-gated channel. Moreover, TPCs recapitulate many of the characteristics of NAADP-induced Ca2+ release i.e. they promote Ca2+ release from acidic stores, correlate with NAADP-binding sites, exhibit a bell-shaped NAADP concentration-response curve, sensitivity to the NAADP antagonist, Ned-19, and provide trigger Ca2+ that is subsequently amplified by ER Ca2+ channels.
Isoforms
There are 3 genes that encode three isoforms of TPC1-3 that differ substantially from each other in their primary sequence (but these differences are preserved across species, such that human and sea urchin TPC1 are more closely related than are human TPC1 and human TPC2). Moreover, the TPC isoforms exhibit different organellar distributions, with TPC1 being found throughout the endo-lysosomal system whereas TPC2 shows a more restricted late-endosomal/lysosomal localization.
Controversy
In spite of a burgeoning literature supporting TPCs as the NAADP-regulated channel, this was challenged in 2012/13 by reports that TPCs are, instead, Na+ channels regulated by the endo-lysosomal lipid, Phosphatidylinositol 3,5-bisphosphate, PI(3,5)P2 and also by metabolic state (via ATP and mTOR).
As a result of this, several groups reinvestigated the permeability properties of TPCs and their role in NAADP-induced Ca2+ release. They agreed that TPCs are indeed permeable to Na+ but they could not necessarily recapitulate the Na+ selectivity shown in the 2012/13 studies. These groups therefore concluded that TPCs were cation channels that conducted both Ca2+ and Na+ (analogous to the NMDA receptor of the plasma membrane).
As for activation by ligands, all groups agree that TPCs are modulated by PI(3,5)P2, though its role is considered by some to be more of a 'permissive' factor rather than an acute signal per se. As for NAADP itself, the conclusion in 2012 that TPCs are not involved in NAADP signalling was partly due to the fact that their transgenic mice (designed to knockout both TPC1 and TPC2; double-knockout, DKO) apparently retained sensitivity to NAADP. However, others have questioned whether these mice are true DKO when they are predicted to retain >90% of the TPC protein sequences (i.e. they express only mildly truncated TPCs which are functional ). In a different DKO mouse that is demonstrably TPC-null, NAADP responses are completely abolished.
On balance, the controversy has been somewhat resolved and it is clear that TPCs are absolutely essential for NAADP. The permeability properties are more equivocal: why some groups observe a Na+ selectivity while others see a mixed Na+/Ca2+ permeability is currently unclear. The necessarily artificial experimental conditions for such as a demanding technique as single-lysosome patch clamp makes it harder to be dogmatic about which ions permeate under native, physiological conditions. It is likely (and the simpler model) that TPCs function as NAADP-gated Ca2+-permeable channels, but it cannot be formally excluded that TPCs, acting as Na+ channels, play a permissive role in a more complex ionic circuit that supports NAADP-induced Ca2+ release [1][2].
Crystal structures of TPC1 from Arabidopsis thaliana reveal the dimeric nature of the molecule, but thus far do not explain the cation selectivity mechanism.
NAADP binding protein
IP3 binds directly to its cognate IP3 receptor which is therefore a true ligand-gated ion channel. In contrast, NAADP may not bind directly to TPCs but may require an intermediate unknown accessory protein. In sea urchin egg homogenate, the binding protein(s) may be smaller than TPCs themselves, judging by photoaffinity labelling with [32P]azido-NAADP. Therefore, the NAADP receptor is likely to be a multi-protein complex on acidic vesicles.
Luminal ions
In addition to NAADP gating the channel, there is evidence that the luminal pH also affects TPC channel activity, either TPC1 [3] or TPC2 [4][5]. However, a clear consensus on the effect of pH has not been reached with some suggesting that acidic pH favours TPC1 or TPC2 opening, whereas others report that a more alkaline pH favours TPC2 opening.
Furthermore, luminal Ca2+ also promotes TPC1 and TPC2 opening (in the latter case, luminal Ca2+ also sensitizes TPCs to NAADP (analogous to luminal Ca2+ regulation of IP3Rs and RyRs), but this demands wider study across isoforms and species. This is one way by which cross-talk can occur between acidic Ca2+ stores and the ER i.e. Ca2+ release from the ER can 'prime' acidic Ca2+ stores and promote further NAADP-dependent Ca2+ responses [6].
Cytosolic ions
To date, the majority of evidence is against the NAADP receptor being regulated by either cytosolic Ca2+ or pH.
NAADP inhibitors
Recently, a selective cell-permeant NAADP antagonist, trans-Ned-19 has been discovered which blocks Ca2+ signals and downstream Ca2+-dependent processes such as differentiation. Prior to that, only high concentrations of blockers of L-type Ca2+ channels (e.g. diltiazem, dihydropyridines) could be used (with obvious concerns over non-NAADP effects).
Although not true antagonism, the NAADP 'receptor' can self-inactivate when bound to non-releasing concentrations of NAADP itself. Such inactivating pre-pulses of NAADP were the first strategy for implicating NAADP in subsequent physiological pathways.
NAADP activators
NAADP is charged and cannot cross cell membranes. Therefore, an inactive, lipophilic ester precursor (NAADP/AM) has been synthesised which crosses membranes and rapidly regenerates NAADP in the cytosol following the action of endogenous esterases.
Caged NAADP is an inactive, membrane-impermeant analog of NAADP that can be introduced into cells by microinjection or a patch pipette. Flash photolysis with a UV light source rapidly converts this into NAADP, allowing the experimenter to precisely manipulate NAADP levels in time and space.
Ca2+ storage
An indirect means of inhibiting NAADP action is to deplete its target Ca2+ stores. As noted above, this usually entails collapsing the H+ gradient with either V-ATPase inhibitors (e.g. Bafilomycin A1) or protonophores (e.g. nigericin or monensin). In platelets it has been suggested that SERCA3 inhibition with tBHQ can also abrogate NAADP-dependent signals.
Transport
The two paralogous enzymes- transmembrane CD38 and GPI anchored CD157, that produce NAADP (and cADPR) in humans both have their active synthesis site in the ectodomain. Though this may involve vesicular synthesis but it has been shown that it is produced at the extracellular sites, and also can act when produced by a different cell or added artificially from outside. So the NAADP has to enter the cell either by diffusion or by transport. Considering the fact that the substrate of NAADP synthesis (NADP) itself is very sparse in the extracellular medium, a purse diffusion based mechanism has been suspected to be less likely than a transporter mediated path. This is compatible with recent data which indicate a carrier mediated transport partially blockable by dipyridamole and cold temperature.