
Specialty medical genetics ICD-9-CM 710.9 DiseasesDB 32101 | ICD-10 Q82.9 OMIM 231070 | |
 | ||
Gerodermia osteodysplastica (GO), also called geroderma osteodysplasticum and Walt Disney dwarfism, is a rare autosomal recessive connective tissue disorder included in the spectrum of cutis laxa syndromes.
Contents
Usage of the name "Walt Disney dwarfism" is attributed to the first known case of the disorder, documented in a 1950 journal report, in which the authors described five affected members from a Swiss family as having the physical appearance of dwarves from a Walt Disney film.
The terms "geroderma" or "gerodermia" can be used interchangeably with "osteodysplastica" or "osteodysplasticum", with the term "hereditaria" sometimes appearing at the end.
Characteristics
Gerodermia osteodysplastica is characterized by symptoms and features which affect the connective tissues, skin and skeletal system.
These are: wrinkly, loose skin over the face, abdomen, and extremites (hands, feet) on the dorsal sides usually worsened by chronic joint laxity and hyperextensibility; fragmented elastic fibers of the skin that are reduced in number, with disorientation of collagen fibers; osteopenia and osteoporosis, with associated fractures; malar hypoplasia (underdeveloped cheek bone), maxillary hypoplasia (underdeveloped upper jaw), mandibular prognathism (protrusion of the lower jaw and chin), bowed long bones, platyspondyly (flattened spine) related to vertebral collapse; kyphoscoliosis (scoliosis with kyphosis, or "hunch back"), metaphyseal peg (an unusual outgrowth of metaphyseal tissue which protrudes into the epiphyseal region of the bone, near the knee); and the overall physical effects and facial appearance of dwarfism with premature aging.
Other features and findings include: intrauterine growth retardation, congenital hip dislocations, winged scapulae (shoulder blades), pes planus (fallen arches), pseudoepiphyses of the second metacarpals (upper bone of the fingers), hypotelorism (close-set eyes), malformed ears, developmental delay, failure to thrive and abnormal electroencephalograph (EEG) readings.
Dental and orthodontal abnormalities in addition to maxillary hypoplasia and mandibular prognathism have also been observed in gerodermia osteodysplastica. Including malocclusion of the dental arches (the maxilla and mandible), radiological findings in some cases have indicated significant overgrowth of the mandibular premolar and molar roots; hypercementosis (overproduction of cementum) of the molars and maxillary incisors; enlarged, funnel-shaped mandibular lingula (spiny structures on the ramus of the mandible); and a radiolucent effect on portions of many teeth, increasing their transparency to x-rays.
Differential diagnosis
Many features of gerodermia osteodysplastica (GO) and another autosomal recessive form of cutis laxa, wrinkly skin syndrome (WSS, Online Mendelian Inheritance in Man (OMIM) 278250), are similar to such an extent that both disorders were believed to be variable phenotypes of a single disorder.
Several delineating factors, however, suggest that gerodermia osteodysplastica and wrinkly skin syndrome are distinct entities, but share the same clinic spectrum.
While the prevailing feature of wrinkly, loose skin is more localized with GO, it is usually systemic, yet eases in severity with age during the course of WSS. Also, as the fontanelles ("soft spots") are usually normal on the heads of infants with GO, they are often enlarged in WSS infants.
Although WSS is associated with mutations of genes on chromosomes 2, 5, 7, 11 and 14; no genetic mutation has been established for GO. A serum sialotransferrin type 2 pattern, also observed with WSS, is not present in GO patients.
But perhaps the most notable feature, differentiating GO from WSS and similar cutis laxa disorders, is the age-specific metaphyseal peg sometimes found in GO-affected long bone, near the knee. Not appearing until around age 4–5, then disappearing by physeal closure, this oddity of bone is thought to represent a specific genetic marker unique to GO and its effects on bone development.
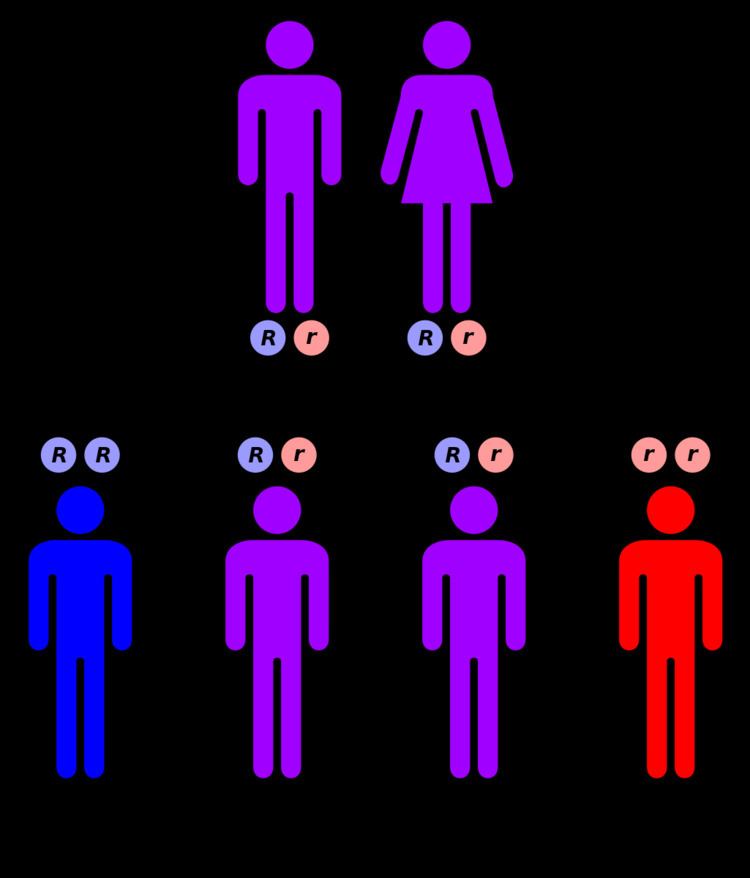
Inheritance
Originally believed to be inherited in an X-linked recessive fashion, gerodermia osteodysplastica is now known to display strictly autosomal recessive inheritance. This means the defective gene responsible for the disorder is located on an autosome, and two copies of the defective gene (one inherited from each parent) are required in order to be born with the disorder. The parents of an individual with an autosomal recessive disorder both carry one copy of the defective gene, but usually do not experience any signs or symptoms of the disorder.
It has been associated with SCYL1BP1.