
 | ||
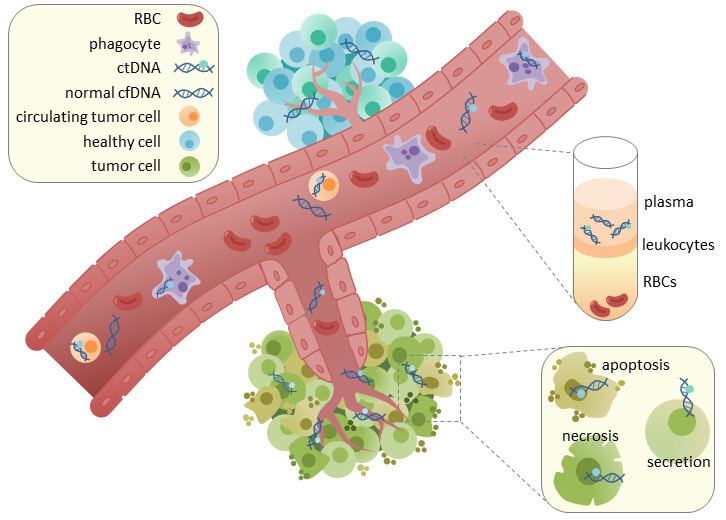
Circulating tumor DNA (ctDNA) is tumor-derived fragmented DNA in the bloodstream that is not associated with cells. ctDNA should not be confused with cell-free DNA (cfDNA), a broader term which describes DNA that is freely circulating in the bloodstream, but is not necessarily of tumor origin. Because ctDNA may reflect the entire tumor genome, it has gained traction for its potential clinical utility; “liquid biopsies” in the form of blood draws may be taken at various time points to monitor tumor progression throughout the treatment regimen.
Contents
- Main advantages
- ctDNA in cancer screening
- ctDNA in cancer monitoring
- ctDNA as a prognostic biomarker
- Cancer research
- Implementation of ctDNA in clinical practice
- Extraction of ctDNA
- Analysis of ctDNA
- Untargeted approaches
- Targeted approaches
- Normal vs tumor DNA detection
- Stability and clearance
- References
ctDNA originates directly from the tumor or from circulating tumor cells (CTCs), which describes viable, intact tumor cells that shed from primary tumors and enter the bloodstream or lymphatic system. The precise mechanism of ctDNA release is unclear. The biological processes postulated to be involved in ctDNA release include apoptosis and necrosis from dying cells, or active release from viable tumor cells. Studies in both human (healthy and cancer patients) and xenografted mice show that the size of fragmented cfDNA is predominantly 166bp long, which corresponds to the length of DNA wrapped around a nucleosome plus a linker. Fragmentation of this length is indicative of apoptotic DNA fragmentation, suggesting that apoptosis may be the primary method of ctDNA release.
In healthy tissue, infiltrating phagocytes are responsible for clearance of apoptotic or necrotic cellular debris, which includes cfDNA. Levels of cfDNA in healthy patients is only present at low levels but detection of higher levels of ctDNA in cancer patients can be detected. This possibly occurs due to inefficient immune cell infiltration to tumor sites, which reduces effective clearance of ctDNA from the bloodstream.
Main advantages
There are several benefits of using ctDNA as a diagnostic or prognostic marker in comparison to other standard methods, such as tissue biopsies. ctDNA represents a real-time biomarker for tumor diagnosis and monitoring, and is useful in situations where collecting a solid tumor sample is too invasive, or if there is not enough sample to genotype. Given the non-invasive nature of ctDNA collection (see ‘Extraction’, below), samples can be collected at multiple time points to monitor tumor progression and response to treatment. Collecting multiple samples to monitor disease progression is not feasible in standard tumor biopsies due to the associated medical risks.
In a study by Gerlinger et al., biopsies were taken from different regions of a primary tumor and metastatic sites. Samples were genotyped and showed both intra- and inter- tumoral heterogeneity. This suggests that a single biopsy may be insufficient to guide clinical action, since it does not encapsulate the entire tumor mass. However, collecting multiple biopsies from different regions of a primary tumor and metastases is invasive and may pose serious medical risks. In such situations, it is possible that ctDNA may present a more practical method of capturing tumor heterogeneity. Recent studies suggest that ctDNA reflects between 0.01%-90% of the tumor, suggesting that the use of ctDNA to survey intra- and inter- tumoral heterogeneity may be beneficial only in certain contexts. Although ctDNA has more potential to capture tumor heterogeneity than traditional tissue biopsies, it is unlikely that ctDNA can provide a complete landscape of tumor heterogeneity and/or metastatic clones.
Notably, ctDNA analysis is not currently used in routine clinical procedures (See ‘Current applications of ctDNA’, below). This may be related to a lack of standardized methods for ctDNA detection - such as extraction, storage, detection, and analysis methods. Furthermore, the use of ctDNA as a tumor biomarker is limited in clinical practice due to a lack of sensitivity/specificity for ctDNA (See ‘Methods’, below).
The applications of ctDNA as a cancer biomarker can be summarized as follows:
- Identification of tumor-specific mutations and detection of tumor heterogeneity in primary and metastatic disease
- Assessment of tumor burden and response to treatment in primary and metastatic disease
- Detection of minimal residual disease (MRD) for early detection of recurrence
- Early detection of primary disease
ctDNA in cancer screening
The clinical utility of ctDNA for the detection of primary disease is in part limited by the sensitivity of current technology; there are low levels of ctDNA present, and driver mutations are unknown. Recently, a team at John Hopkins assessed the ability to detect ctDNA using digital PCR in 640 patients with various types of cancer. ctDNA was detected in only 48% to 73% of cases, suggesting that ctDNA may be a poor method for early cancer detection, since not all patients had detectable levels of ctDNA. Similar findings were observed by Heitzer et al., who analyzed ctDNA from colorectal patients. Importantly, in a study conducted by Kopreski et al., mutations in KRAS, an oncogene commonly mutated in colorectal cancer, were detected in the colorectal tissue of otherwise clinically healthy individuals. In fact, 58% of individuals with detectable KRAS mutations showed no evidence of neoplasia on colonoscopy, which may be indicative of false positivity. However, because no follow-up studies were pursued within this cohort, it is possible that these represent premalignant lesions. Taken together, these results suggest that the use of ctDNA as a biomarker in the context of cancer screening is limited both by sensitivity and specificity.
ctDNA in cancer monitoring
While ctDNA may currently not be useful in a cancer diagnosis or early screening, it may be more amenable to survey ctDNA to monitor disease progression and treatment response once a formal clinical diagnosis has been made. Prior sequencing of the tumor biopsy may also provide known mutations for disease-specific, targeted sequencing of ctDNA.
For most solid tumors, imaging methods such as CT, PET and MRI are considered the “gold standard” for disease monitoring. However, these methods are expensive and are limited in detection capability; there is poor resolution of tumors of <1 cm in size. Of note, monitoring of certain cancers is also done through analysis of protein biomarkers. For example, prostate-specific antigen (PSA) can be monitored by collecting blood samples from prostate cancer patients. However, these markers are often unreliable, and suitable protein biomarkers are absent for many cancers. Therefore, ctDNA may provide greater utility for monitoring tumor burden for a greater range of cancer types, including breast cancer, periampullary cancer, colorectal cancer, head and neck cancer, melanoma, renal cancer and prostate cancer.
Another appeal of ctDNA is that it has the potential to offer “real-time” tracking of tumor evolution; taking multiple samples throughout a treatment time-course (before, during and after treatment regimens) may yield insight regarding treatment efficacy and response, which can guide subsequent therapeutic strategies. The genetic aberrations that lead to treatment resistance or colonization to distal sites are not likely to be represented in a primary tumor biopsy that has been taken before a treatment regimen is initiated. Therefore, ctDNA represents an attractive alternative to detect treatment resistant or metastatic clones.
For example, in a study conducted by Spindler et al., KRAS/BRAF mutations from plasma samples were monitored in 108 patients with metastatic colorectal cancer before and after cycles of the chemotherapeutic drugs cetuximab and irinotecan. The concentration of KRAS mutations from plasma samples decreased with successive cycles of treatment, and loss of detectable mutations was associated with treatment benefit. Remarkably, researchers also observed the appearance of new mutations, which may be representative of acquired resistance. This study provides evidence that ctDNA may be a suitable biomarker for monitoring colorectal cancer progression.
Evidence of disease by traditional imaging methods, such as CT, PET or MRI may be absent after tumor resection. Therefore, ctDNA analysis poses a potential avenue to detect minimal residual disease (MRD), and thus the possibility of tumor recurrence, in cases where bulk tumor is absent by conventional imaging methods. A comparison of MRD detection by CT imaging compared to ctDNA has been previously done in individuals with stage II colon cancer; in this study, researchers were able to detect ctDNA in individuals who showed no sign of clinical malignancy by a CT scan, suggesting that ctDNA detection has greater sensitivity to assess MRD. However, the authors acknowledge that ctDNA analysis is not without limitations; plasma samples collected post-operatively were only able to predict recurrence at 36 months in 48% of cases.
ctDNA as a prognostic biomarker
The use of ctDNA as a prognostic biomarker has been previously described in cervical cancer, colorectal cancer, pancreatic cancer, and melanoma. For example, in a study of KRAS mutations and CDKN2A hypermethylation in individuals with colorectal cancer, researchers found a 100% 2-year survival rate in individuals with ctDNA levels below detectable limit. Similar findings were observed by Diehl et al., who found that patients with detectable ctDNA tended to relapse within 1 year. This suggests that there may be prognostic utility of ctDNA levels in colorectal cancer.
One of the more reliable prognostic indicators is assessment of disease stage. Generally, low stage cancers are associated with a more favourable prognosis. Although some studies have shown a trend of higher ctDNA levels in patients with metastatic cancer, it is important to note that ctDNA burden does not always correlate with traditional cancer staging. For this reason, it is unlikely that ctDNA will replace current methods for stratifying patients based on less-aggressive or more-aggressive disease; current methods to assess tumor stage and grade are multi-factorial and include histological, immunohistochemical, and molecular analysis. Additionally, in the case of breast cancer and colorectal cancer, there are FDA-approved assays to differentiate between high-risk and low-risk cancers from tissue biopsy samples. While it is unlikely that ctDNA will be of clinical utility as a sole predictor of prognosis, it may be beneficial in cases where a tissue biopsy is not possible, or archived tumor samples are not available for genetic analysis.
Cancer research
The emergence of drug-resistant tumors due to intra- and inter-tumoral heterogeneity an issue in treatment efficacy. A minor genetic clone within the tumor can expand after treatment if it carries a drug-resistant mutation. Initial biopsies can miss these clones due to low frequency or spatial separation of cells within the tumor. For example, since a biopsy only samples a small part of the tumor, clones that resides in a different location may go unnoticed. This can mislead research that focuses on studying the role of tumor heterogeneity in cancer progression and relapse. The use of ctDNA in research can alleviate these concerns because it could provide a more representative 'screenshot' of the genetic diversity of cancer at both primary and metastatic sites. For example, ctDNA has been shown to be useful in studying the clonal evolution of a patient’s cancer before and after treatment regimens.
Implementation of ctDNA in clinical practice
While ctDNA analysis is not a routine clinical procedure, a ctDNA assay to detect EGFR mutations in non-small cell lung cancer (NSCLC) was recently approved based on a 2014 phase IV clinical trial which focused on the efficacy of gefitinib in NSCLC patients with an EGFR mutation. The study included matched tumor and plasma samples and determined that ctDNA analysis may be used where tumor samples are not available for mutational analysis. Similar findings were made in a study profiling somatic mutations in solid tumors from 105 patients enrolled in phase I clinical trials for molecular targeted agents. In this study, researchers found that ctDNA analysis was beneficial in cases where repeated tumor biopsies were not feasible.
Implementation of ctDNA in clinical practice is largely hindered by the lack of standardized methods for ctDNA processing and analysis. Standardization of methods for sample collection (including time of collection), downstream processing (DNA extraction and amplification), quantification and validation must be established before ctDNA analysis can become a routine clinical assay. Furthermore, creation of a panel of ‘standard’ tumor-associated biomarkers may be necessary given the resolution of current ctDNA sequencing and detection methods. Sequencing tumor-specific aberrations from plasma samples may also help exclude contaminating cfDNA from analysis; elevated levels of cfDNA from normal cells may be attributed to non-cancer related causes.
Extraction of ctDNA
The main appeal of ctDNA analysis is that it is extracted in a non-invasive manner through blood collection. Acquisition of cfDNA or ctDNA typically requires collection of approximately 3mL of blood into EDTA-coated tubes. The use of EDTA is important to reduce coagulation of blood. The plasma and serum fractions of blood can be separated through a centrifugation step. ctDNA or cfDNA can be subsequently extracted from these fractions. Although serum tends to have greater levels of cfDNA, this is primarily attributed to DNA from lymphocytes. High levels of contaminating cfDNA is sub-optimal because this can decrease the sensitivity of ctDNA detection. Therefore, the majority of studies use plasma for ctDNA isolation. Plasma is then processed again by centrifugation to remove residual intact blood cells. The supernatant is used for DNA extraction, which can be performed using commercially available kits.
Analysis of ctDNA
The analysis of ctDNA after extraction requires the use of various amplification and sequencing methods. These methods can be separated into two main groups based on whether the goal is to interrogate all genes in an untargeted approach, or if the goal is to monitor specific genes and mutations in a targeted approach.
Untargeted approaches
A whole genome or whole exome sequencing approaches may be necessary to discover new mutations in tumor DNA while monitoring disease burden or tracking drug resistance. Untargeted approaches are also useful in research to observe tumor heterogeneity or to discover new drug targets. However, while untargeted methods may be necessary in certain applications, it is more expensive and has lower resolution. This makes it difficult to detect rare mutations, or in situations where low ctDNA levels are present (such as minimal residual disease). Furthermore, there can be problems distinguishing between DNA from tumor cells and DNA from normal cells using a whole genome approach.
Whole genome or exome sequencing typically use high throughput DNA sequencing technologies. Limiting the sequencing to only the whole exome instead can decrease expense and increase speed, but at the cost of losing information about mutations in the non-coding regulatory regions of DNA. While simply looking at DNA polymorphisms through sequencing does not differentiate DNA from tumor or normal cells, this problem can be resolved by comparing against a control sample of normal DNA (for example, DNA obtained through a buccal swab.) Importantly, whole genome and whole exome sequencing are useful for initial mutation discovery. This provides information for the use of more sensitive targeted techniques, which can then be used for disease monitoring purposes.
Digital Karyotyping
Unlike normal karyotyping where a dye is used to stain chromosomal bands in order to visualize the chromosomes, digital karyotyping uses DNA sequences of loci throughout the genome in order to calculate copy number variation. Copy number variations are common in cancers and describe situations where loss of heterozygosity of a gene may lead to decreased function due to lower expression, or duplication of a gene, which leads to overexpression.
Personalized analysis of rearranged ends (PARE)
After the whole genome is sequenced using a high throughput sequencing method, such as Illumina HiSeq, PARE is applied to the data to analyze chromosomal rearrangements and translocations. This technique was originally designed to analyze solid tumor DNA but was modified for ctDNA applications.
DNA Methylation
Proper epigenetic marking is essential for normal gene expression and cell function and aberrant alterations in epigenetic patterns is a hallmark of cancer. A normal epigenetic status is maintained in a cell at least in part through DNA methylation. Measuring aberrant methylation patterns in ctDNA is a possible due to stable methylation of regions of DNA referred to as “CpG islands”. Methylation of ctDNA can be detected through bisulfite treatment. Bisulfite treatment chemically converts unmethylated cytosines into a uracil while leaving methylated cytosines unmodified. DNA is subsequently sequenced, and any alterations to the DNA methylation pattern can be identified.
Targeted approaches
In a targeted approach, sequencing of ctDNA can be directed towards a genetic panel constructed based on mutational hotspots for the cancer of interest. This is especially important for informing treatment in situations where mutations are identified in druggable targets. Personalizing targeted analysis of ctDNA to each patient is also possible by combining liquid biopsies with standard primary tissue biopsies. Whole genome or whole exome sequencing of the primary tumor biopsy allows for discovery of genetic mutations specific to a patient’s tumor, and can be used for subsequent targeted sequencing of the patient’s ctDNA. The highest sensitivity of ctDNA detection is accomplished through targeted sequencing of specific single nucleotide polymorphisms (SNPs). Commonly mutated genes, such as oncogenes, which typically have hotspot mutations, are good candidates for targeted sequencing approaches. Conversely, most tumor suppressor genes have a wide array of possible loss of function mutations throughout the gene, and as such are not suitable for targeted sequencing.
Targeted approaches have the advantage of amplifying ctDNA through polymerase chain reactions (PCR) or digital PCR. This is especially important when analyzing ctDNA not only because there are relatively low levels of DNA circulating in the bloodstream, but also because ctDNA makes up a small proportion of the total cell-free DNA extracted. Therefore, amplification of regions of interest can drastically improve sensitivity of ctDNA detection. However, amplification through PCR can introduce errors given the inherent error rate of DNA polymerases. Errors introduced during sequencing can also decrease the sensitivity of detecting ctDNA mutations.
Digital droplet PCR (ddPCR)
Digital droplet PCR utilizes a droplet generator to partition single pieces of DNA into droplets using an oil/water emulsion. Then individual polymerase chain reactions occur in each droplet using selected primers against regions of ctDNA and proceeds to endpoint. The presence of the sequences of interest is measured by fluorescent probes, which bind to the amplified region. ddPCR allows for highly quantitative assessment of allele and mutant frequencies in ctDNA but is limited by the number of fluorescent probes that can be used in one assay (up to 5). The sensitivity of the assay can vary depending on the amount of DNA analyzed and is around 1 in 10,000.
Beads, Emulsification, Amplification, and Magnetics (BEAMing)
This technique builds upon digital droplet PCR in order to identify mutations in ctDNA using flow cytometry. After ctDNA is extracted from blood, PCR is performed with primers designed to target the regions of interest. These primers also contain specific DNA sequences, or tags. The amplified DNA is mixed with streptavidin-coated magnetic beads and emulsified into droplets. Biotinylated primers designed to bind to the tags are used to amplify the DNA. Biotinylation allows the amplified DNA to bind to the magnetic beads, which are coated with streptavidin. After the PCR is complete, the DNA-bound beads are separated using a magnet. The DNA on the beads are then denatured and allowed to hybridize with fluorescent oligonucleotides specific to each DNA template. The resulting bead-DNA complexes are then analyzed using flow cytometry. This technique is able to capture allele and mutation frequencies due to coupling with ddPCR. However, unlike with ddPCR, a larger number of DNA sequences can be interrogated due to the flexibility of using fluorescently bound probes. Another advantage of this system is that the DNA isolated can also be used for downstream sequencing. Sensitivity is 1.6 in 10,000 to 4.3 in 100,000.
CAncer Personalized Profiling by deep Sequencing (CAPP-Seq)
This technique uses biotinylated oligonucleotide selector probes to target sequences of DNA relevant to ctDNA detection. Publicly available cancer databases were used to construct a library of probes against recurrent mutations in cancer by calculating their recurrence index. The protocol was optimized for the low DNA levels observed in ctDNA collection. Then the isolated DNA undergoes deep sequencing for increased sensitivity. This technique allows for the interrogation of hundreds of DNA regions. The ctDNA detection sensitivity of CAPP-Seq is reported to be 1 molecule in 10,000.
Tagged AMplicon deep Sequencing (TAM-Seq)
TAM-Seq allows targeted sequencing of entire genes to detect mutations in ctDNA. First a general amplification step is performed using primers that span the entire gene of interest in 150-200bp sections. Then, a microfluidics system is used to attached adaptors with a unique identifier to each amplicon to further amplify the DNA in parallel singleplex reactions. This technique was shown to successfully identify mutations scattered in the TP53 tumor suppressor gene in advanced ovarian cancer patients. The sensitivity of this technique is 1 in 50.
Safe-Sequencing (Safe-Seq)
Safe-Seq decreases the error rate of massively parallel sequencing in order to increase the sensitivity to rare mutants. It achieves this by addition of a unique identifier (UID) sequence to each DNA template. The DNA is then amplified using the added UIDs and sequenced. All DNA molecules with the same UID (a UID family) should have the same reported DNA sequence since they were amplified from one molecule. However, mutations can be introduced through amplification, or incorrect base assignments may be called in the sequencing and analysis steps. The presence of the UID allows these methodology errors to be separated from true mutations of the ctDNA. A mutation is considered a ‘supermutant’ if 95% of the sequenced reads are in agreement. The sensitivity of this approach is 9 in 1 million.
Duplex sequencing
This method is an improvement on the single UIDs added in the Safe-Seq technique. In duplex sequencing, randomized double-stranded DNA act as unique tags and are attached to an invariant spacer. Tags are attached to both ends of a DNA fragment (α and β tags), which results in two unique templates for PCR - one strand with an α tag on the 5’ end and a β tag on the 3’ end and the other strand with a β tag on the 5’ end and an α tag on the 3’ end. These DNA fragments are then amplified with primers against the invariant sequences of the tags. The amplified DNA is sequenced and analyzed. DNA with the duplex adaptors are compared and mutations are only accepted if there is a consensus between both strands. This method takes into account both errors from sequencing and errors from early stage PCR amplification. The sensitivity of the approach to discovering mutants is 1 in 107.
Integrated Digital Error Suppression (iDES)-enhanced CAPP-Seq
iDES improves CAPP-Seq analysis of ctDNA in order to decrease error and therefore increase sensitivity of detection. Reported in 2016, iDES combines CAPP-Seq with duplex barcoding sequencing technology and with a computational algorithm that removes stereotypical errors associated with the CAPP-Seq hybridization step. The sensitivity of this improved version of CAPP-Seq is 4 in 100,000 copies.
“Normal” vs tumor DNA detection
An important consideration of using ctDNA as a cancer biomarker is the ability to distinguish between cfDNA from normal versus malignant cells. cfDNA is released by non-malignant cells during normal cellular turnover, but also during procedures such as surgery, radiotherapy, or chemotherapy. It is thought that lymphocytes are the primary contributors to cfDNA in serum. Notably, some studies have shown that increased levels of cfDNA are found in cancer patients compared to healthy individuals, and that in some contexts, cfDNA levels correlate with disease progression. Similarly, Koffler et al. have found a higher concordance of cfDNA in diseased individuals compared to a healthy population. However, it is difficult to determine the proportion of DNA that is of tumor-origin, since there are other physiological circumstances where cfDNA is elevated (for example, during inflammation or injury). For this reason, targeted assays to detect microsatellite instability or copy number variation, point mutations, and loss of heterozygosity of certain known cancer related genes may be more beneficial than bulk sequencing of cfDNA.
Stability and clearance
Not much is known on the stability of ctDNA in the bloodstream but it is thought to be relatively short. Free ctDNA may be quickly degraded by the spleen, kidneys or liver. Although clearance dynamics have not been specifically studied for ctDNA in cancer, research based on cell-free fetal DNA was shown to be cleared in a biphasic manner with half-lives of 1 hour and 13 hours respectively. In the context of liquid biopsies, this relatively quick turnover would be beneficial for obtaining accurate 'snapshots' of disease progression.