
EC number 1.14.13.32 | Entrez 1576 | |
 | ||
Aliases CYP3A4, CP33, CP34, CYP3A, CYP3A3, CYPIIIA3, CYPIIIA4, HLP, NF-25, P450C3, P450PCN1, cytochrome P450 family 3 subfamily A member 4 External IDs OMIM: 124010 HomoloGene: 111391 GeneCards: CYP3A4 | ||
Cytochrome P450 3A4 (abbreviated CYP3A4) (EC 1.14.13.97), is an important enzyme in the body, mainly found in the liver and in the intestine. It oxidizes small foreign organic molecules (xenobiotics), such as toxins or drugs, so that they can be removed from the body.
Contents
- Function
- Evolution
- Tissue distribution
- Mechanisms
- Inhibition through fruit ingestion
- Variability
- Induction
- Turnover
- Technology
- CYP3A4 ligands
- Interactive pathway map
- References
While many drugs are deactivated by CYP3A4, there are also some drugs which are activated by the enzyme. Some substances, such as grapefruit juice and some drugs, interfere with the action of CYP3A4. These substances will therefore either amplify or weaken the action of those drugs that are modified by CYP3A4.
CYP3A4 is a member of the cytochrome P450 family of oxidizing enzymes. Several other members of this family are also involved in drug metabolism, but CYP3A4 is the most common and the most versatile one. Like all members of this family, it is a hemoprotein, i.e. a protein containing a heme group with an iron atom. In humans, the CYP3A4 protein is encoded by the CYP3A4 gene. This gene is part of a cluster of cytochrome P450 genes on chromosome 7q21.1.
Function
CYP3A4 is a member of the cytochrome P450 superfamily of enzymes. The cytochrome P450 proteins are monooxygenases that catalyze many reactions involved in drug metabolism and synthesis of cholesterol, steroids, and other lipids components.
The CYP3A4 protein localizes to the endoplasmic reticulum, and its expression is induced by glucocorticoids and some pharmacological agents. This enzyme is involved in the metabolism of approximately half the drugs that are used today, including acetaminophen, codeine, ciclosporin (cyclosporin), diazepam, and erythromycin. The enzyme also metabolizes some steroids and carcinogens. Most drugs undergo deactivation by CYP3A4, either directly or by facilitated excretion from the body. Also, many substances are bioactivated by CYP3A4 to form their active compounds, and many protoxins being toxicated into their toxic forms (for examples - see table below).
CYP3A4 also possesses epoxygenase activity in that it metabolizes arachidonic acid to epoxyeicosatrienoic acids (EETs), i.e. (±)-8,9-, (±)-11,12-, and (±)-14,15-epoxyeicosatrienoic acids. The EETs have a wide range of activities including the promotion of certain types of cancers (see epoxyeicosatetraenoic acid#cancer). CYP3A4 promotes the growth of various types of human cancer cell lines in culture by producing (±)-14,15-epoxyeicosatrienoic acids which stimulate these cells to grow, The cytochrome P450 is also reported to have fatty acid monooxgenase activity for metabolizing arachidonic acid to 20-Hydroxyeicosatetraenoic acid (20-HETE). 20-HETE has a wide range of activities that also include growth stimulation in breast and other types of cancers (see 12-hydroxyeicosatetraenoic acid#cancer).
Evolution
The CYP3A4 gene exhibits a much more complicated upstream regulatory region in comparison with its paralogs. This increased complexity renders the CYP3A4 gene more sensitive to endogenous and exogenous PXR and CAR ligands, instead of relying on gene variants for wider specificity. Chimpanzee and human CYP3A4 are highly conserved in metabolism of many ligands, although four amino acids positively selected in humans led to a 5-fold benzylation of 7-BFC in the presence of the hepatotoxic secondary bile acid lithocholic acid. This change in consequence contributes to an increased human defense against cholestasis.
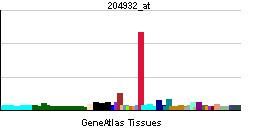
Tissue distribution
Fetuses do not really express CYP3A4 in their liver tissue, but rather CYP3A7 (EC 1.14.14.1), which acts on a similar range of substrates. CYP3A4 is absent in fetal liver but increases to approximately 40% of adult levels in the fourth month of life and 72% at 12 months.
Although CYP3A4 is predominantly found in the liver, it is also present in other organs and tissues of the body, where it may play an important role in metabolism. CYP3A4 in the intestine plays an important role in the metabolism of certain drugs. Often this allows prodrugs to be activated and absorbed - as in the case of the histamine H1-receptor antagonist terfenadine.
Recently CYP3A4 has also been identified in the brain, however its role in the central nervous system is still unknown.
Mechanisms
Cytochrome P450 enzymes perform an assortment of modifications on a variety of ligands, utilizing its large active site and its ability to bind more than one substrate at a time to perform complicated chemical alterations in the metabolism of endogenous and exogenous compounds. These include hydroxylation, epoxidation of olefins, aromatic oxidation, heteroatom oxidations, N- and O- dealkylation reactions, aldehyde oxidations, dehydrogenation reactions, and aromatase activity.
Hydroxylation of an sp3 C-H bond is one of the ways in which CYP3A4 (and cytochrome P450 oxygenases) affects its ligand. In fact, hydroxylation is sometimes followed by dehydrogenation, leading to more complex metabolites. An example of a molecule that undergoes more than one reaction due to CYP3A4 includes tamoxifen, which is hydroxylated to 4-hydroxy-tamoxifen and then dehydrated to 4-hydroxy-tamoxifen quinone methide. Two mechanisms have been proposed as the primary pathway of hydroxylation in P450 enzymes.
The first pathway suggested is a cage-controlled radical method (“oxygen rebound”), and the second involves a concerted mechanism that does not utilize a radical intermediate but instead acts very quickly via a “radical clock”.
Inhibition through fruit ingestion
In 1998, various researchers showed that grapefruit juice, and grapefruit in general, is a potent inhibitor of CYP3A4, which can affect the metabolism of a variety of drugs, increasing their bioavailability. In some cases, this can lead to a fatal interaction with drugs like astemizole or terfenadine. The effect of grapefruit juice with regard to drug absorption was originally discovered in 1989. The first published report on grapefruit drug interactions was in 1991 in the Lancet entitled "Interactions of Citrus Juices with Felodipine and Nifedipine" and was the first reported food-drug interaction clinically. The effects of grapefruit last from 3–7 days, with the greatest effects when taken simultaneously with the drug.
In addition to grapefruit, other fruits have similar effects. Noni (M. citrifolia), for example, is a dietary supplement typically consumed as a juice and also inhibits CYP3A4; pomegranate juice has this effect as well.
Variability
While over 28 single nucleotide polymorphisms (SNPs) have been identified in the CYP3A4 gene, it has been found that this does not translate into significant interindividual variability in vivo. It can be supposed that this may be due to the induction of CYP3A4 on exposure to substrates.
CYP3A4 alleles which have been reported to have minimal function compared to wild-type include CYP3A4*6 (an A17776 insertion) and CYP3A4*17 (F189S). Both of these SNPs led to decreased catalytic activity with certain ligands, including testosterone and nifedipine in comparison to wild-type metabolism.
Variability in CYP3A4 function can be determined noninvasively by the erythromycin breath test (ERMBT). The ERMBT estimates in vivo CYP3A4 activity by measuring the radiolabelled carbon dioxide exhaled after an intravenous dose of (14C-N-methyl)-erythromycin.
Induction
CYP3A4 is induced by a wide variety of ligands. These ligands bind to the pregnane X receptor (PXR). The activated PXR complex forms a heterodimer with the retinoid X receptor (RXR), which binds to the XREM region of the CYP3A4 gene. XREM is a regulatory region of the CYP3A4 gene, and binding causes a cooperative interaction with proximal promoter regions of the gene, resulting in increased transcription and expression of CYP3A4. Activation of the PXR/RXR heterodimer initiates transcription of the CYP3A4 promoter region and gene. Ligand binding increases when in the presence of CYP3A4 ligands, such as in the presence of aflatoxin B1, M1, and G1. Indeed, due to the enzyme’s large and malleable active site, it is possible for the enzyme to bind multiple ligands at once, leading to potentially detrimental side effects.
Induction of CYP3A4 has been shown to vary in humans depending on sex. Evidence shows an increased drug clearance by CYP3A4 in women, even when accounting for differences in body weight. A study by Wobold et al. (2003) found that the median CYP3A4 levels measured from surgically removed liver samples of a random sample of women exceeded CYP3A4 levels in the livers of men by 129%. CYP3A4 mRNA transcripts were found in similar proportions, suggesting a pre-translational mechanism for the up-regulation of CYP3A4 in women. The exact cause of this elevated level of enzyme in women is still under speculation, however studies have elucidated other mechanisms (such as CYP3A5 or CYP3A7 compensation for lowered levels of CYP3A4) that affect drug clearance in both men and women.
CYP3A4 substrate activation varies amongst different animal species. Certain ligands activate human PXR, which promotes CYP3A4 transcription, while showing no activation in other species. For instance, mouse PXR is not activated by rifampicin and human PXR is not activated by pregnenalone 16α-carbonitrile In order to facilitate study of CYP3A4 functional pathways in vivo, mouse strains have been developed using transgenes in order to produce null/human CYP3A4 and PXR crosses. Although humanized hCYP3A4 mice successfully expressed the enzyme in their intestinal tract, low levels of hCYP3A4 were found in the liver. This effect has been attributed to CYP3A4 regulation by the growth hormone signal transduction pathway. In addition to providing an in vivo model, humanized CYP3A4 mice (hCYP3A4) have been used to further emphasize gender differences in CYP3A4 activity.
CYP3A4 activity levels have also been linked to diet and environmental factors, such as duration of exposure to xenobiotic substances. Due to the enzyme’s extensive presence in the intestinal mucosa, the enzyme has shown sensitivity to starvation symptoms and is upregulated in defense of adverse effects. Indeed, in fatheaded minnows, unfed female fish were shown to have increased PXR and CYP3A4 expression, and displayed a more pronounced response to xenobiotic factors after exposure after several days of starvation. By studying animal models and keeping in mind the innate differences in CYP3A4 activation, investigators can better predict drug metabolism and side effects in human CYP3A4 pathways.
Turnover
Estimates of the turnover rate of human CYP3A4 vary widely. For hepatic CYP3A4, in vivo methods yield estimates of enzyme half-life mainly in the range of 70 to 140 hours, whereas in vitro methods give estimates from 26 to 79 hours. Turnover of gut CYP3A4 is likely to be a function of the rate of enterocyte renewal; an indirect approach based on recovery of activity following exposure to grapefruit juice yields measurements in the 12- to 33-hour range.
Technology
Due to membrane-bound CYP3A4’s natural propensity to conglomerate, it has historically been difficult to study drug binding in both solution and on surfaces. Co-crystallization is difficult since the substrates tend to have a low Kd (between 5-150 μM) and low solubility in aqueous solutions. A successful strategy in isolating the bound enzyme is the functional stabilization of monomeric CYP3A4 on Ag nanoparticles produced from nanosphere lithography and analyzed via localized surface plasmon resonance spectroscopy (LSPR). These analyses can be used as a high-sensitivity assay of drug binding, and may become integral in further high-throughput assays utilized in initial drug discovery testing. In addition to LSPR, CYP3A4-Nanodisc complexes have been found helpful in other applications including solid-state NMR, redox potentiometry, and steady-state enzyme kinetics.
CYP3A4 ligands
Following is a table of selected substrates, inducers and inhibitors of CYP3A4. Where classes of agents are listed, there may be exceptions within the class.
Inhibitors of CYP3A4 can be classified by their potency, such as:
Interactive pathway map
Click on genes, proteins and metabolites below to link to respective articles.