
Entrez 54658 | Ensembl ENSG00000241635 | |
 | ||
Aliases UGT1A1, BILIQTL1, GNT1, HUG-BR1, UDPGT, UDPGT 1-1, UGT1, UGT1A, UDP glucuronosyltransferase family 1 member A1 External IDs OMIM: 191740 MGI: 98898 HomoloGene: 128034 GeneCards: UGT1A1 | ||
UDP-glucuronosyltransferase 1-1 also known as UGT-1A is an enzyme that in humans is encoded by the UGT1A1 gene.
Contents
UGT-1A is a uridine diphosphate glucuronosyltransferase (UDP-glucuronosyltransferase, UDPGT), an enzyme of the glucuronidation pathway that transforms small lipophilic molecules, such as steroids, bilirubin, hormones, and drugs, into water-soluble, excretable metabolites.
Gene
The UGT1A1 gene is part of a complex locus that encodes several UDP-glucuronosyltransferases. The locus includes thirteen unique alternate first exons followed by four common exons. Four of the alternate first exons are considered pseudogenes. Each of the remaining nine 5' exons may be spliced to the four common exons, resulting in nine proteins with different N-termini and identical C-termini. Each first exon encodes the substrate binding site, and is regulated by its own promoter. Over 100 genetic variants within the UGT1A1 gene have been described, some of which confer increased, reduced or inactive enzymatic activity. The UGT nomenclature committee has compiled a list of these variants, naming each with a * symbol followed by a number.
Clinical significance
Mutations in this gene cause serious problems for bilirubin metabolism; each syndrome can be caused by one or many mutations, so they are differentiated mostly by symptoms and not particular mutations:
Pharmacogenetics
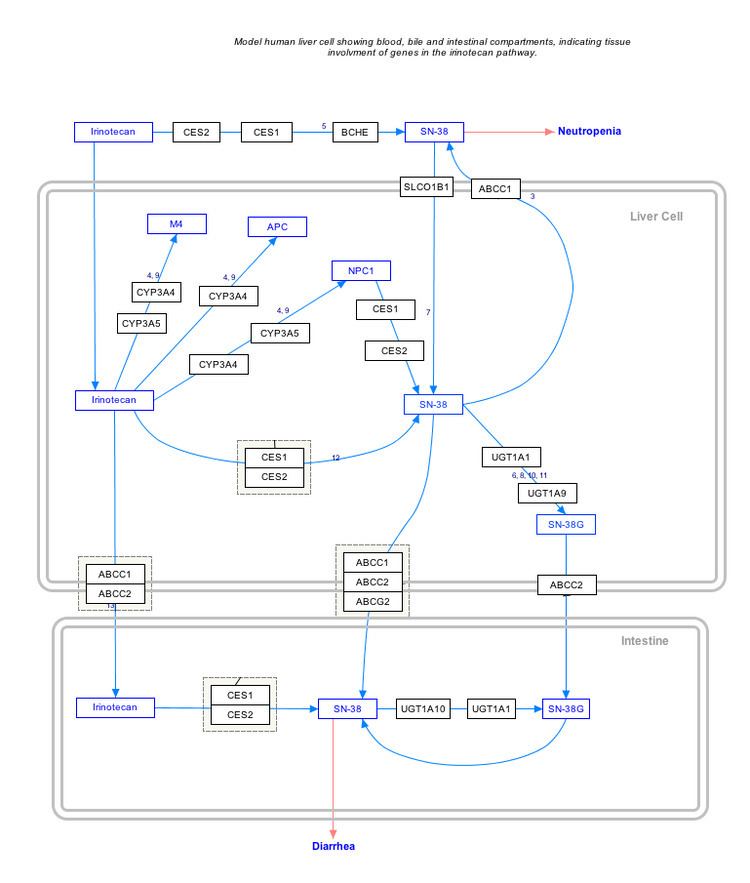
Genetic variations within the UGT1A1 gene have also been associated with the development of certain drug toxicities. The UGT1A1*28 variant, the same allele behind many cases of Gilbert syndrome, has been associated with an increased risk for neutropenia in patients receiving the chemotherapeutic drug irinotecan, and the U.S. Food and Drug Administration recommends on the irinotecan drug label that patients with the *28/*28 genotype receive a lower starting dose of the drug. The *28 allele has also shown associations with an increased risk for developing diarrhea in patients receiving irinotecan. The UGT1A1*6 variant, more common in Asian populations than the *28 variant, has also shown associations with the development of irinotecan toxicities. Patients who are heterozygous or homozygous for the *6 allele may have a higher risk for developing neutropenia and diarrhea as compared to those with the UGT1A1*1/*1 genotype.
Interactive pathway map
Click on genes, proteins and metabolites below to link to respective articles.