
 | ||
Predictive genomics is at the intersection of multiple disciplines: predictive medicine, personal genomics and translational bioinformatics. Specifically, predictive genomics deals with the future phenotypic outcomes via prediction in areas such as complex multifactorial diseases in humans. To date, the success of predictive genomics has been dependent on the genetic framework underlying these applications, typically explored in genome-wide association (GWA) studies. The identification of associated single-nucleotide polymorphisms (variation of a DNA sequence in a population) underpin GWA studies in complex diseases that have ranged from Type 2 Diabetes (T2D), Age-related macular degeneration (AMD) and Crohn’s Disease.
Contents
- Objectives
- Identify associated variants to disease
- Translation research to clinical
- Individualising healthcare
- Applications in complex human diseases
- Age related macular degeneration
- Type 2 diabetes
- Celiac disease
- Limitations
- Variants in prediction SNPs and alternatives
- Interacting variants higher order analysis
- Population size and scope
- Other applications
- References
Although the Human Genome Project has progressively improved the fidelity of sequence determination, the overbearing complexity of the genome hinders the identification of associated or ultimately causal variants. In particular, there are likely to be a large number of implicated genetic loci which exhibit small marginal effects.
Objectives
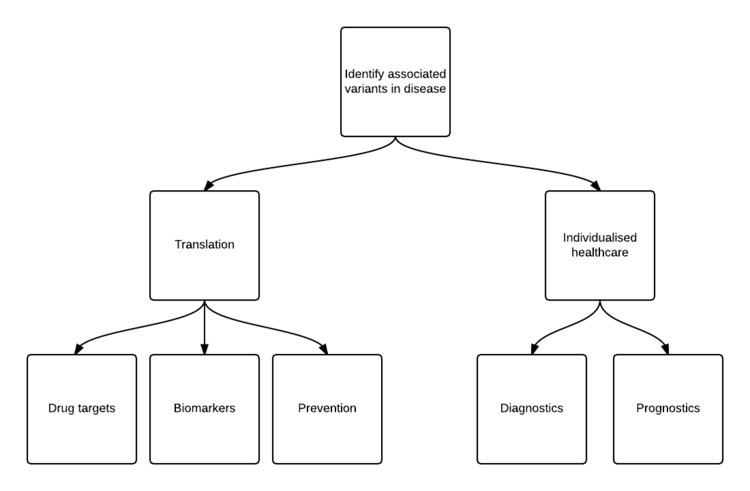
A number of short- and long-term goals exist for predictive genomics. The identification of associated variants underpin all other downstream endeavors that point toward better data-cum-knowledge outcomes. In particular, those outcomes that facilitate clinical improvement and individualised healthcare further lead to actionable measures in diagnosis, prognosis and prevention.
Identify associated variants to disease
Whilst the single-gene, single-disease hypothesis holds for Mendelian disorders such as Huntington's disease and Cystic Fibrosis, complex diseases and traits are affected by a number of gene loci and genetic variants with varying risk. A precursor to the development of preventative, prognostic and diagnostic tools in these diseases requires mapping genetic loci in disease etiology and discovering causal mutations. Creating a ‘genomic profile’ of individuals with the number of variants at the genome-wide level facilitates not only the prediction of disease prior to onset, but also serves as a primer to increasing the knowledge of causal variants.
The foremost difficulty in achieving this goal is to understand the functionality of these variants with respect to areas of physiological and molecular importance in conjunction with phenotype. If associated variants are mapped to sequences with unknown function, then this restricts the ability for specific targeting in areas of interest. Therefore the ability for predictive genomics to succeed also depends upon other related areas such as the functional annotation of the genome (ENCODE).
Translation: research to clinical
The identification of causal variants, genes and pathways leads to opportunities that bridge the divide between research and clinical usage. If successful, the subsequent discovery of therapeutic targets within implicated biological pathways have consequences for both treatment and prevention. Furthermore, the downstream effect of identifying disease-relevant biomarkers allow for improvements to monitoring disease progression and response-to-treatment, where the implementation of these results into clinical decision support systems (CDSS) facilitate personalised medicine and outcomes. Even if only marginally effectual, the repeated replication of associated variants can offer significant translational value.
Individualising healthcare
The significance of translation from research to clinical usage relates to use of the complete knowledge of an individual to develop personalised approaches to disease management. The caveat with this is that there have been difficulties in both prediction and inference for complex diseases. Therefore, unless individuals have an overwhelming high or low number of risk alleles, there is a limit to the predictive accuracy of their ‘genomic profiles’. However, preliminary examples of predictive genomics for personalising healthcare include: using an individuals gene expression data to monitor progress to treatment, or using the genomic profile of the P450 drug metabolising system of individuals to assist dosage and selection.
Applications in complex human diseases
In the table below is a performance comparison of diseases selected on disease frequency and known heritability estimates, with use of single-nucleotide polymorphism (SNP) based models reflecting known genetic factors for a European population (subject to change as more associations are discovered).
In the applications of predictive genomics below, these complex diseases either lack or are lacking reliable diagnostics for disease. Given the medical consequences of these diseases, the economic impact is also significant. However, none of the use cases below has been translated into the clinic.
Age-related macular degeneration
Age-related macular degeneration (AMD) is one of the flagship complex diseases from the genomic revolution with over 19 associated genetic loci replicated in GWA studies. In particular, the first significant genetic risk variant was identified in the complement factor H(CFH) gene in 2005 motivating the search for more genetic variants in the disease. Over the past decade, a number of models have been proposed to assess individual risk to AMD. The genetic predisposition of AMD risk varies from 45% to 71% where highly effectual odds ratios (OR) have been reported (greater than 2.0 per allele in some cases). In 2013, a comprehensive case-control GWA study with approximately 77,000 observations involving 18 international research groups from the International AMD Genetics Consortium implicated 19 gene loci and 9 biological pathways including the regulation of complement, lipid metabolism and angiogenic activity. The predictive performance of the full model including all 19 loci exhibited 0.74 AUC - according to Jakobsdottir et al., 0.75 AUC is sufficient to distinguish between extreme cases and controls. In particular, of the 19 associated gene loci, there were 7 newly discovered loci, which the authors point to as additional entry points into AMD etiology and drug targets.
Type 2 diabetes
Type 2 diabetes (T2D), an extremely common metabolic disorder, has demonstrated interplay between many environmental and genetic risk factors leading to disease onset. A number of risk assessment models incorporating a number of demographic, environmental and clinical risk factors are already shown to elicit reasonable discrimination in case-control studies; it has been proposed that identifying genetic variants that contribute to T2D as for standalone prediction or in conjunction with current risk models can improve prediction of T2D risk, if current models lack sufficient coverage of the full effect of an individual's genotype. Approximately 20 associated SNPs have been replicated in T2D; however, their effect sizes do not seem to be substantial: OR 1.37 for SNPs in the TCF7L2 gene purported to give highest genetic risk.
In 2009, a study was conducted on the WTCCC (GWA study involving 7 cohorts with 7 diseases: including bipolar disorder, Crohn’s disease, hypertension, rheumatoid arthritis, Type I Diabetes (T1D) and Type II Diabetes (T2D)). With particular attention to T2D, Evans et al. were able to discern a marginal increase in AUC (+0.04) based on genome-wide information with respect to known susceptible variants. However, non-genetic based tests such as the Cambridge and Framingham offspring risk scores have been purported to perform better than genetic-risk models with 20 loci. Moreover, the addition of genetic risk with these phenotypical models did not produce statistically significant AUC results.
Celiac disease
Celiac disease (CD) is a complex immune disorder that has been found to have strong genetic links in disease. In particular, human leukocyte antigen (HLA) genes are strongly implicated in CD development and HLA testing is undertaken in clinical practice. However, although there are serological and histological tests available for CD, these clinical screenings have been found to generate false positives. In 2014, Abraham et al. used a genomic risk score (GRS) generated over 6 cohorts with an AUC of 0.86 to 0.90.
Limitations
For predictive genomics to address their objectives, there must be an improvement in the accuracy of prediction through added methods or improvements to current techniques and to demonstrate that there is bonafide improvement in patient outcomes. Currently, although AUC (Area Under the ROC) is the de facto metric in comparing and evaluating the performance of predictive models, there is no consensus as to what kind of score is sufficient for clinical use. Jakobsdottir et al. states that 0.75 AUC is sufficient for discriminating between clear cases and controls; however, this is still arbitrary. The positive predictive value (PPV) must be high enough to avoid a higher prevalence of false-positives.
Variants in prediction: SNPs and alternatives
SNPs identified in GWA studies are considered to give better predictive performance if they have high effect sizes of Odds Ratios (OR). A case study involving 5 use cases of genomic prediction demonstrate that SNPs with extremely small p-values, and by implication extreme OR do not give extreme differences in discrimination. They point out that use of significantly associated genetic variants does not necessarily lead to better classification. Alternatively, CNV (copy number variants) have been proposed to usurp SNPs as better candidates for prediction with BMI stratified across different ethnicities demonstrating better, although marginal, improvement of CNVs over SNPs for prediction. Furthermore, a comparison of over 10 complex disorders in prediction with respect to family history and SNPs for prediction did not suggest better discrimination with SNPs.
Interacting variants: higher order analysis
Currently, the prevailing standard of risk models focus on univariate analysis rather than focusing upon interactions of higher order. Therefore although typical GWA studies are able to detect a number of statistically significant loci, they have not been sufficient to fully explain the estimates of theoretical genetic heritability. It has been demonstrated by Goudey et al. that both 2-way and 3-way interactions between SNPs are able to explain trait variance relative to single SNPs. Goudey et al. also states that the barrier to expansion of higher order interactions has been limited by the intractability of exhaustive search techniques (see NP-complete).
Population: size and scope
The issues surrounding sample size and number of variants become exacerbated particularly when GWA studies consider variants of volume in the order of millions. Therefore, due to the current constraints in the curse of dimensionality, prior screening methods that decrease the number of loci to below the number of observations may be used before modelling disease risk. Hayes et al. states that population size must be >100,000 in order to achieve high accuracy under their model assumptions; the exception is the case where there is a small effective population size. Furthermore, ethnic specific GWA studies show that each group has varied detectability of variants in terms of: frequency, linkage disequilibrium – the co-inheritance of SNPs through generations – and the actual loci themselves.
Other applications
Predictive genomics has not been constrained to prediction of complex diseases. For instance, Hayes et al. uses genomic prediction for livestock, crop and forage species selection, where predicted results are currently in use. Furthermore, Kambouris et al. discusses the use of ‘genomic profiles’ for the performance of elite athletes noting individualised and personalised training regimens for both dietary and physical aspects. Additionally, Kayser et al. point to DNA profiling in forensics as a beneficiary of the genomic revolution.