
Start date 1990 Period 1990 – 2003 | Completed April 2003 | |
 | ||
Awards Albany Medical Center Prize | ||
The Human Genome Project (HGP) was an international scientific research project with the goal of determining the sequence of nucleotide base pairs that make up human DNA, and of identifying and mapping all of the genes of the human genome from both a physical and a functional standpoint. It remains the world's largest collaborative biological project. After the idea was picked up in 1984 by the US government when the planning started, the project formally launched in 1990 and was declared complete in 2003. Funding came from the US government through the National Institutes of Health (NIH) as well as numerous other groups from around the world. A parallel project was conducted outside of government by the Celera Corporation, or Celera Genomics, which was formally launched in 1998. Most of the government-sponsored sequencing was performed in twenty universities and research centers in the United States, the United Kingdom, Japan, France, Germany, Canada, and China.
Contents
- History
- State of completion
- Applications and proposed benefits
- Techniques and analysis
- Findings
- Accomplishment
- Public versus private approaches
- Genome donors
- Developments
- Ethical legal and social issues
- References

The Human Genome Project originally aimed to map the nucleotides contained in a human haploid reference genome (more than three billion). The "genome" of any given individual is unique; mapping the "human genome" involved sequencing a small number of individuals and then assembling these together to get a complete sequence for each chromosome. The finished human genome is thus a mosaic, not representing any one individual.

History

The Human Genome Project was a 13-year-long, publicly funded project initiated in 1990 with the objective of determining the DNA sequence of the entire euchromatic human genome within 15 years. In May 1985, Robert Sinsheimer organized a workshop to discuss sequencing the human genome, but for a number of reasons the NIH was uninterested in pursuing the proposal. The following March, the Santa Fe Workshop was organized by Charles DeLisi and David Smith of the Department of Energy's Office of Health and Environmental Research (OHER). At the same time Renato Dulbecco proposed whole genome sequencing in an essay in Science. James Watson followed two months later with a workshop held at the Cold Spring Harbor Laboratory.
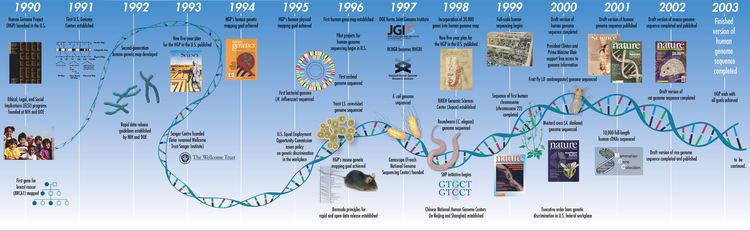
The fact that the Santa Fe workshop was motivated and supported by a Federal Agency opened a path, albeit a difficult and tortuous one, for converting the idea into public policy. In a memo to the Assistant Secretary for Energy Research (Alvin Trivelpiece), Charles DeLisi, who was then Director of OHER, outlined a broad plan for the project. This started a long and complex chain of events which led to approved reprogramming of funds that enabled OHER to launch the Project in 1986, and to recommend the first line item for the HGP, which was in President Reagan's 1988 budget submission, and ultimately approved by the Congress. Of particular importance in Congressional approval was the advocacy of Senator Peter Domenici, whom DeLisi had befriended. Domenici chaired the Senate Committee on Energy and Natural Resources, as well as the Budget Committee, both of which were key in the DOE budget process. Congress added a comparable amount to the NIH budget, thereby beginning official funding by both agencies.

Alvin Trivelpiece sought and obtained the approval of DeLisi's proposal by Deputy Secretary William Flynn Martin. This chart was used in the spring of 1986 by Trivelpiece, then Director of the Office of Energy Research in the Department of Energy, to brief Martin and Under Secretary Joseph Salgado regarding his intention to reprogram $4 million to initiate the project with the approval of Secretary Herrington. This reprogramming was followed by a line item budget of $16 million in the Reagan Administration’s 1987 budget submission to Congress. It subsequently passed both Houses. The Project was planned for 15 years.
Candidate technologies were already being considered for the proposed undertaking at least as early as 1985.
In 1990, the two major funding agencies, DOE and NIH, developed a memorandum of understanding in order to coordinate plans and set the clock for the initiation of the Project to 1990. At that time, David Galas was Director of the renamed “Office of Biological and Environmental Research” in the U.S. Department of Energy’s Office of Science and James Watson headed the NIH Genome Program. In 1993, Aristides Patrinos succeeded Galas and Francis Collins succeeded James Watson, assuming the role of overall Project Head as Director of the U.S. National Institutes of Health (NIH) National Center for Human Genome Research (which would later become the National Human Genome Research Institute). A working draft of the genome was announced in 2000 and the papers describing it were published in February 2001. A more complete draft was published in 2003, and genome "finishing" work continued for more than a decade.
The $3-billion project was formally founded in 1990 by the US Department of Energy and the National Institutes of Health, and was expected to take 15 years. In addition to the United States, the international consortium comprised geneticists in the United Kingdom, France, Australia, China and myriad other spontaneous relationships.
Due to widespread international cooperation and advances in the field of genomics (especially in sequence analysis), as well as major advances in computing technology, a 'rough draft' of the genome was finished in 2000 (announced jointly by U.S. President Bill Clinton and the British Prime Minister Tony Blair on June 26, 2000). This first available rough draft assembly of the genome was completed by the Genome Bioinformatics Group at the University of California, Santa Cruz, primarily led by then graduate student Jim Kent. Ongoing sequencing led to the announcement of the essentially complete genome on April 14, 2003, two years earlier than planned. In May 2006, another milestone was passed on the way to completion of the project, when the sequence of the last chromosome was published in Nature.
State of completion
The project was not able to sequence all the DNA found in human cells. It sequenced only "euchromatic" regions of the genome, which make up more than 95% of the genome. The other regions, called "heterochromatic" are found in centromeres and telomeres, and were not sequenced under the project.
The Human Genome Project was declared complete in April 2003. An initial rough draft of the human genome was available in June 2000 and by February 2001 a working draft had been completed and published followed by the final sequencing mapping of the human genome on April 14, 2003. Although this was reported to cover 99% of the euchromatic human genome with 99.99% accuracy, a major quality assessment of the human genome sequence was published on May 27, 2004 indicating over 92% of sampling exceeded 99.99% accuracy which was within the intended goal. Further analyses and papers on the HGP continue to occur.
Applications and proposed benefits
The sequencing of the human genome holds benefits for many fields, from molecular medicine to human evolution. The Human Genome Project, through its sequencing of the DNA, can help us understand diseases including: genotyping of specific viruses to direct appropriate treatment; identification of mutations linked to different forms of cancer; the design of medication and more accurate prediction of their effects; advancement in forensic applied sciences; biofuels and other energy applications; agriculture, animal husbandry, bioprocessing; risk assessment; bioarcheology, anthropology and evolution. Another proposed benefit is the commercial development of genomics research related to DNA based products, a multibillion-dollar industry.
The sequence of the DNA is stored in databases available to anyone on the Internet. The U.S. National Center for Biotechnology Information (and sister organizations in Europe and Japan) house the gene sequence in a database known as GenBank, along with sequences of known and hypothetical genes and proteins. Other organizations, such as the UCSC Genome Browser at the University of California, Santa Cruz, and Ensembl present additional data and annotation and powerful tools for visualizing and searching it. Computer programs have been developed to analyze the data, because the data itself is difficult to interpret without such programs. Generally speaking, advances in genome sequencing technology have followed Moore’s Law, a concept from computer science which states that integrated circuits can increase in complexity at an exponential rate. This means that the speeds at which whole genomes can be sequenced can increase at a similar rate, as was seen during the development of the above-mentioned Human Genome Project.
Techniques and analysis
The process of identifying the boundaries between genes and other features in a raw DNA sequence is called genome annotation and is in the domain of bioinformatics. While expert biologists make the best annotators, their work proceeds slowly, and computer programs are increasingly used to meet the high-throughput demands of genome sequencing projects. Beginning in 2008, a new technology known as RNA-seq was introduced that allowed scientists to directly sequence the messenger RNA in cells. This replaced previous methods of annotation, which relied on inherent properties of the DNA sequence, with direct measurement, which was much more accurate. Today, annotation of the human genome and other genomes relies primarily on deep sequencing of the transcripts in every human tissue using RNA-seq. These experiments have revealed that over 90% of genes contain at least one and usually several alternative splice variants, in which the exons are combined in different ways to produce 2 or more gene products from the same locus.
The genome published by the HGP does not represent the sequence of every individual's genome. It is the combined mosaic of a small number of anonymous donors, all of European origin. The HGP genome is a scaffold for future work in identifying differences among individuals. Subsequent projects sequenced the genomes of multiple distinct ethnic groups, though as of today there is still only one "reference genome."
Findings
Key findings of the draft (2001) and complete (2004) genome sequences include:
- There are approximately 22,300 protein-coding genes in human beings, the same range as in other mammals.
- The human genome has significantly more segmental duplications (nearly identical, repeated sections of DNA) than had been previously suspected.
- At the time when the draft sequence was published fewer than 7% of protein families appeared to be vertebrate specific.
Accomplishment
The Human Genome Project was started in 1990 with the goal of sequencing and identifying all three billion chemical units in the human genetic instruction set, finding the genetic roots of disease and then developing treatments. It is considered a Mega Project because the human genome has approximately 3.3 billion base-pairs. With the sequence in hand, the next step was to identify the genetic variants that increase the risk for common diseases like cancer and diabetes.
It was far too expensive at that time to think of sequencing patients’ whole genomes. So the National Institutes of Health embraced the idea for a "shortcut", which was to look just at sites on the genome where many people have a variant DNA unit. The theory behind the shortcut was that, since the major diseases are common, so too would be the genetic variants that caused them. Natural selection keeps the human genome free of variants that damage health before children are grown, the theory held, but fails against variants that strike later in life, allowing them to become quite common. (In 2002 the National Institutes of Health started a $138 million dollar project called the HapMap to catalog the common variants in European, East Asian and African genomes.)
The genome was broken into smaller pieces; approximately 150,000 base pairs in length. These pieces were then ligated into a type of vector known as "bacterial artificial chromosomes", or BACs, which are derived from bacterial chromosomes which have been genetically engineered. The vectors containing the genes can be inserted into bacteria where they are copied by the bacterial DNA replication machinery. Each of these pieces was then sequenced separately as a small "shotgun" project and then assembled. The larger, 150,000 base pairs go together to create chromosomes. This is known as the "hierarchical shotgun" approach, because the genome is first broken into relatively large chunks, which are then mapped to chromosomes before being selected for sequencing.
Funding came from the US government through the National Institutes of Health in the United States, and a UK charity organization, the Wellcome Trust, as well as numerous other groups from around the world. The funding supported a number of large sequencing centers including those at Whitehead Institute, the Sanger Centre, Washington University in St. Louis, and Baylor College of Medicine.
The United Nations Educational, Scientific and Cultural Organization (UNESCO) served as an important channel for the involvement of developing countries in the Human Genome Project.
Public versus private approaches
In 1998, a similar, privately funded quest was launched by the American researcher Craig Venter, and his firm Celera Genomics. Venter was a scientist at the NIH during the early 1990s when the project was initiated. The $300,000,000 Celera effort was intended to proceed at a faster pace and at a fraction of the cost of the roughly $3 billion publicly funded project. The Celera approach was able to proceed at a much more rapid rate, and at a lower cost than the public project because it relied upon data made available by the publicly funded project.
Celera used a technique called whole genome shotgun sequencing, employing pairwise end sequencing, which had been used to sequence bacterial genomes of up to six million base pairs in length, but not for anything nearly as large as the three billion base pair human genome.
Celera initially announced that it would seek patent protection on "only 200–300" genes, but later amended this to seeking "intellectual property protection" on "fully-characterized important structures" amounting to 100–300 targets. The firm eventually filed preliminary ("place-holder") patent applications on 6,500 whole or partial genes. Celera also promised to publish their findings in accordance with the terms of the 1996 "Bermuda Statement", by releasing new data annually (the HGP released its new data daily), although, unlike the publicly funded project, they would not permit free redistribution or scientific use of the data. The publicly funded competitors were compelled to release the first draft of the human genome before Celera for this reason. On July 7, 2000, the UCSC Genome Bioinformatics Group released a first working draft on the web. The scientific community downloaded about 500 GB of information from the UCSC genome server in the first 24 hours of free and unrestricted access.
In March 2000, President Clinton announced that the genome sequence could not be patented, and should be made freely available to all researchers. The statement sent Celera's stock plummeting and dragged down the biotechnology-heavy Nasdaq. The biotechnology sector lost about $50 billion in market capitalization in two days.
Although the working draft was announced in June 2000, it was not until February 2001 that Celera and the HGP scientists published details of their drafts. Special issues of Nature (which published the publicly funded project's scientific paper) and Science (which published Celera's paper) described the methods used to produce the draft sequence and offered analysis of the sequence. These drafts covered about 83% of the genome (90% of the euchromatic regions with 150,000 gaps and the order and orientation of many segments not yet established). In February 2001, at the time of the joint publications, press releases announced that the project had been completed by both groups. Improved drafts were announced in 2003 and 2005, filling in to approximately 92% of the sequence currently.
Genome donors
In the IHGSC international public-sector HGP, researchers collected blood (female) or sperm (male) samples from a large number of donors. Only a few of many collected samples were processed as DNA resources. Thus the donor identities were protected so neither donors nor scientists could know whose DNA was sequenced. DNA clones from many different libraries were used in the overall project, with most of those libraries being created by Pieter J. de Jong's. Much of the sequence (>70%) of the reference genome produced by the public HGP came from a single anonymous male donor from Buffalo, New York (code name RP11).
HGP scientists used white blood cells from the blood of two male and two female donors (randomly selected from 20 of each) – each donor yielding a separate DNA library. One of these libraries (RP11) was used considerably more than others, due to quality considerations. One minor technical issue is that male samples contain just over half as much DNA from the sex chromosomes (one X chromosome and one Y chromosome) compared to female samples (which contain two X chromosomes). The other 22 chromosomes (the autosomes) are the same for both sexes.
Although the main sequencing phase of the HGP has been completed, studies of DNA variation continue in the International HapMap Project, whose goal is to identify patterns of single-nucleotide polymorphism (SNP) groups (called haplotypes, or “haps”). The DNA samples for the HapMap came from a total of 270 individuals: Yoruba people in Ibadan, Nigeria; Japanese people in Tokyo; Han Chinese in Beijing; and the French Centre d’Etude du Polymorphisme Humain (CEPH) resource, which consisted of residents of the United States having ancestry from Western and Northern Europe.
In the Celera Genomics private-sector project, DNA from five different individuals were used for sequencing. The lead scientist of Celera Genomics at that time, Craig Venter, later acknowledged (in a public letter to the journal Science) that his DNA was one of 21 samples in the pool, five of which were selected for use.
In 2007, a team led by Jonathan Rothberg published James Watson's entire genome, unveiling the six-billion-nucleotide genome of a single individual for the first time.
Developments
The work on interpretation and analysis of genome data is still in its initial stages. It is anticipated that detailed knowledge of the human genome will provide new avenues for advances in medicine and biotechnology. Clear practical results of the project emerged even before the work was finished. For example, a number of companies, such as Myriad Genetics, started offering easy ways to administer genetic tests that can show predisposition to a variety of illnesses, including breast cancer, hemostasis disorders, cystic fibrosis, liver diseases and many others. Also, the etiologies for cancers, Alzheimer's disease and other areas of clinical interest are considered likely to benefit from genome information and possibly may lead in the long term to significant advances in their management.
There are also many tangible benefits for biologists. For example, a researcher investigating a certain form of cancer may have narrowed down his/her search to a particular gene. By visiting the human genome database on the World Wide Web, this researcher can examine what other scientists have written about this gene, including (potentially) the three-dimensional structure of its product, its function(s), its evolutionary relationships to other human genes, or to genes in mice or yeast or fruit flies, possible detrimental mutations, interactions with other genes, body tissues in which this gene is activated, and diseases associated with this gene or other datatypes. Further, deeper understanding of the disease processes at the level of molecular biology may determine new therapeutic procedures. Given the established importance of DNA in molecular biology and its central role in determining the fundamental operation of cellular processes, it is likely that expanded knowledge in this area will facilitate medical advances in numerous areas of clinical interest that may not have been possible without them.
The analysis of similarities between DNA sequences from different organisms is also opening new avenues in the study of evolution. In many cases, evolutionary questions can now be framed in terms of molecular biology; indeed, many major evolutionary milestones (the emergence of the ribosome and organelles, the development of embryos with body plans, the vertebrate immune system) can be related to the molecular level. Many questions about the similarities and differences between humans and our closest relatives (the primates, and indeed the other mammals) are expected to be illuminated by the data in this project.
The project inspired and paved the way for genomic work in other fields, such as agriculture. For example, by studying the genetic composition of Tritium aestivum, the world’s most commonly used bread wheat, great insight has been gained into the ways that domestication has impacted the evolution of the plant. Which loci are most susceptible to manipulation, and how does this play out in evolutionary terms? Genetic sequencing has allowed these questions to be addressed for the first time, as specific loci can be compared in wild and domesticated strains of the plant. This will allow for advances in genetic modification in the future which could yield healthier, more disease-resistant wheat crops.
Ethical, legal and social issues
At the onset of the Human Genome Project several ethical, legal, and social concerns were raised in regards to how increased knowledge of the human genome could be used to discriminate against people. One of the main concerns of most individuals was the fear that both employers and health insurance companies would refuse to hire individuals or refuse to provide insurance to people because of a health concern indicated by someone's genes. In 1996 the United States passed the Health Insurance Portability and Accountability Act (HIPAA) which protects against the unauthorized and non-consensual release of individually identifiable health information to any entity not actively engaged in the provision of healthcare services to a patient.
Along with identifying all of the approximately 20,000–25,000 genes in the human genome, the Human Genome Project also sought to address the ethical, legal, and social issues that were created by the onset of the project. For that the Ethical, Legal, and Social Implications (ELSI) program was founded in 1990. Five percent of the annual budget was allocated to address the ELSI arising from the project. This budget started at approximately $1.57 million in the year 1990, but increased to approximately $18 million in the year 2014.
Whilst the project may offer significant benefits to medicine and scientific research, some authors have emphasized the need to address the potential social consequences of mapping the human genome. "Molecularising disease and their possible cure will have a profound impact on what patients expect from medical help and the new generation of doctors' perception of illness."