
Entrez 5184 | Ensembl ENSG00000124299 | |
 | ||
Aliases PEPD, PROLIDASE, peptidase D External IDs MGI: 97542 HomoloGene: 239 GeneCards: PEPD | ||
Xaa-Pro dipeptidase, also known as prolidase, is an enzyme that in humans is encoded by the PEPD gene.
Contents
Function
Xaa-Pro dipeptidase is a cytosolic dipeptidase that hydrolyzes dipeptides with proline or hydroxyproline at the carboxy terminus (but not Pro-Pro). It is important in collagen metabolism because of the high levels of imino acids. Mutations at the PEPD locus cause prolidase deficiency. This is characterised by Iminodipeptidurea, skin ulcers, mental retardation and recurrent infections.
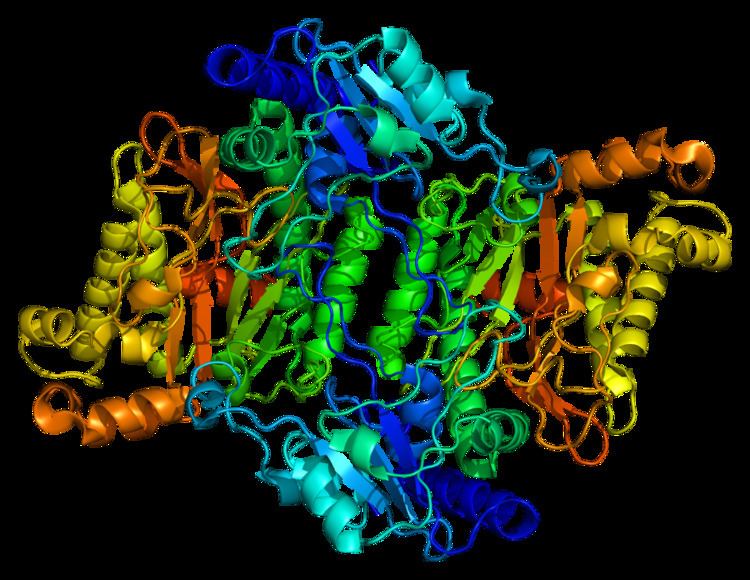
Structure
Prolidases fall under a subclass of metallopeptidases that involve binuclear active site metal clusters. This metal cluster facilitates catalysis by serving as a substrate binding site, activating nucleophiles, and stabilizing the transition state. Furthermore, prolidases are classified under a smaller family called “pita-bread” enzymes, which cleave amido-, imido-, and amidino- containing bonds. The “pita-bread” fold, containing a metal center flanked by two well-defined substrate binding pockets enabled prolidase to specifically cleave between any non-proline amino acid and proline.
The first ever solved structure of prolidase came from the hyperthermophilic archaeon Pyrococcus furiosus (Pfprol). This dimer has a crystal structure shows two approximately symmetrical monomers that both have an N-terminal domain, made up of a six-stranded mixed β-sheet flanked by five α-helices, a helical linker, and C-terminal domain, consisting of a mixed six-stranded β-sheet flanked by four α-helices. The curved β-sheet of Domain II has a “pita-bread” fold. The active site lies on the inner surface of the β-sheet of Domain II, with a notable dinuclear Co cluster anchored by the side chains of two aspartate residues (Asp209 and Asp220), two glutamate residues (Glu313 and Glu327), and a histidine residue (His284). Carboxylate groups of aspartate and glutamine residues serve as bridges between the two Co atoms. In the crystallization process, the Co atoms are replaced with Zn, which hinders enzymatic activity.
Unlike Pfprol, the structure of the human variant remains poorly understood. Sequence homology between human and Pfprol yield only 25% identity and 43% similarity. The two available structures of human prolidase available on the Protein Data Bank are homodimers contain either Na or Mn, which bind to similar amino acids as those in Pfprol: Glu412 (Glu313 in Pfprol), binds to the first ion, Asp276 (Asp209 in Pfprol) binds to the second ion, and Asp287 and Glu452 bind to both (Asp220 and Glu327 in Pfprol).
Function
Due to proline’s cyclic structure, only few peptidases could cleave the bond between proline and other amino acids. Along with prolinase, prolidase are the only known enzymes that can break down dipeptides to yield free proline. Prolidase serve to hydrolyze both dietary and endogenous Xaa-Pro dipeptides. More specifically, it is essential in catalyzing the last step of the degradation of procollagen, collagen, and other proline-containing peptides into free amino acids to be used for cellular growth. Additionally, it also participates in the process of recycling proline from Xaa-Pro dipeptides for collagen resynthesis. Proline and hydroyxyproline make up a quarter of the amino acid residues in collegen, which is the most abundant protein in the body by mass and plays an important role in maintaining connective tissue in the body.
Mechanism
The mechanism for prolidase catalytic activity remains largely uncharacterized. However, biochemical and structural analyses of aminopeptidase (APPro), methionine aminopeptidase (MetAP), and prolidase, all members of the “pita-bread” metalloenzymes, suggest that they share a common mechanism scheme. The main difference arises in the location of the carbonyl oxygen atom of the scissile peptide bond.
The following mechanism shows a proposed scheme for a metal-dependent “pita-bread” enzyme with residue numbering corresponding to those found in methionine aminopeptidase from E. coli. As shown in Intermediate I of the figure, three potential acidic amino acid residues interact with the N-terminus of the substrate in a fashion that is yet to be determined. The carbonyl and amide groups of the scissile peptide bond interact with the first metal ion, M1, in addition to His178 and His79, respectively. M1 and Glu204 activate a water molecule to prepare it nucleophilic attack at the carbonyl carbon of the scissile peptide bond. Then, the tetrahedral intermediate (Intermediate II) becomes stabilized from interactions with M1 and His178. Lastly, Glu204 donates a proton to the amine of the leaving peptide (P1’). This leads to the breakdown of the intermediate (Intermediate III), which retains its interactions with M1 and His178.
Regulation
Post-translational modifications of prolidase regulate its enzymatic abilities. Phosphorylation of prolidase has been shown to increase its activity while dephosphorylation leads to a decrease in enzyme activity. Analysis of known consensus sequence required for serine/threonine phosphorylation revealed that prolidase contains at least three potential sites for serine/threonine phosphorylation. Nitric oxide, both exogenously acquired and endogenously generated, was shown to increase prolidase activity in a time- and dose-dependent manner via phosphorylation at these serine and threonine sites. Additionally, prolidase may also be regulated at tyrosine phosphorylation sites, which are mediated by FAK and MAPK signaling pathways.
Disease relevance
Deficiency in prolidase leads to a rare, severe autosomal recessive disorder (prolidase deficiency) that causes many chronic, debilitating health conditions in humans. These phenotypical symptoms vary and may include skin ulcerations, mental retardation, splenomegaly, recurrent infections, photosensitivity, hyperkeratosis, and unusual facial appearance. Furthermore, prolidase activity was found to be abnormal compared to healthy levels in various medical conditions including but limited to: bipolar disorder, breast cancer, endometrial cancer, keloid scar formation, erectile dysfunction, liver disease, lung cancer, hypertension, melanoma, and chronic pancreatitis. In some cancers with increased levels of prolidase activity, such as melanoma, the differential expression of prolidase and its substrate specificity for dipeptides with proline at the carboxyl end suggests the potential of prolidase in becoming a viable, selective endogenous enzyme target for proline prodrugs. Serum prolidase enzyme activity is also currently being explored as a possible, reliable marker for diseases including chronic hepatitis B and liver fibrosis.
Other applications
Decontamination: Prolidase from the hyperthermophilic archaeon Pyrococcus furiosus (Pfprol) shows potential for application in decontamination of organophosphorus nerve agents in chemical warfare agents. Additionally, prolidase could also serve to detect fluorine-containing organophosphorus neurotoxins, like the G-type chemical warfare agents, and could antagonize organophosphorous intoxication and protect against the effects of diisopropylfluorophosphate when encapsulated in liposomes.
Model organisms
Model organisms have been used in the study of PEPD function. A conditional knockout mouse line called Pepdtm1a(KOMP)Wtsi was generated at the Wellcome Trust Sanger Institute. Male and female animals underwent a standardized phenotypic screen to determine the effects of deletion. Additional screens performed: - In-depth immunological phenotyping