
Species Human Entrez 5243 | Ensembl ENSG00000085563 | |
 | ||
Aliases ABCB1, ABC20, CD243, CLCS, GP170, MDR1, P-GP, PGY1, ATP binding cassette subfamily B member 1 External IDs | ||
P-glycoprotein 1 (permeability glycoprotein, abbreviated as P-gp or Pgp) also known as multidrug resistance protein 1 (MDR1) or ATP-binding cassette sub-family B member 1 (ABCB1) or cluster of differentiation 243 (CD243) is an important protein of the cell membrane that pumps many foreign substances out of cells. More formally, it is an ATP-dependent efflux pump with broad substrate specificity. It exists in animals, fungi and bacteria and likely evolved as a defense mechanism against harmful substances.
Contents
- Function
- Structure
- Mechanism of action
- Tissue distribution
- Exogenous inhibitors
- Detecting the activity of the transporter
- History
- References
P-gp is extensively distributed and expressed in the intestinal epithelium where it pumps xenobiotics (such as toxins or drugs) back into the intestinal lumen, in liver cells where it pumps them into bile ducts, in the cells of the proximal tubule of the kidney where it pumps them into urine-conducting ducts, and in the capillary endothelial cells composing the blood–brain barrier and blood-testis barrier, where it pumps them back into the capillaries.
P-gp is a glycoprotein that in humans is encoded by the ABCB1 gene. P-gp is a well-characterized ABC-transporter (which transports a wide variety of substrates across extra- and intracellular membranes) of the MDR/TAP subfamily. The normal excretion of xenobiotics back into the gut lumen by P-gp pharmacokinetically reduces the efficacy of some pharmaceutical drugs (which are said to be P-gp substrates). In addition, some cancer cells also express large amounts of P-gp, further amplifying that effect and rendering these cancers multidrug resistant. Many drugs inhibit P-gp, typically incidentally rather than as their main mechanism of action; some foods do as well. Any such substance can sometimes be called a P-gp inhibitor.
P-gp was discovered in 1971 by Victor Ling.
Function
The protein belongs to the superfamily of ATP-binding cassette (ABC) transporters. ABC proteins transport various molecules across extra- and intra-cellular membranes. ABC genes are divided into seven distinct subfamilies (ABC1, MDR/TAP, MRP, ALD, OABP, GCN20, White). This protein is a member of the MDR/TAP subfamily. Members of the MDR/TAP subfamily are involved in multidrug resistance. P-gp is an ATP-dependent drug efflux pump for xenobiotic compounds with broad substrate specificity. It is responsible for decreased drug accumulation in multidrug-resistant cells and often mediates the development of resistance to anticancer drugs. This protein also functions as a transporter in the blood–brain barrier.
P-gp transports various substrates across the cell membrane including:
Its ability to transport the above substrates accounts for the many roles of P-gp including:
It is inhibited by many drugs, such as Amiodarone, Azithromycin, Captopril, Clarithromycin, Cyclosporine, Piperine, Quercetin, Quinidine, Quinine, Reserpine, Ritonavir, Tariquidar, and Verapamil.
Structure
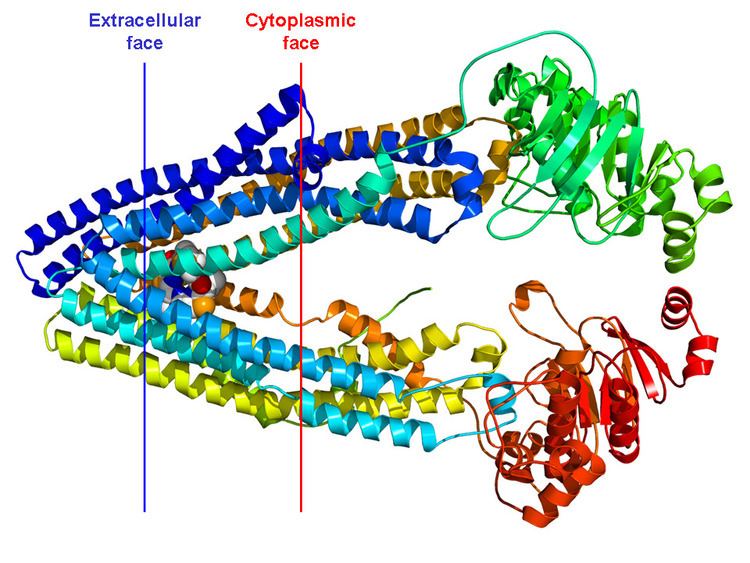
P-gp is a 170 kDa transmembrane glycoprotein, which includes 10-15 kDa of N-terminal glycosylation. The N-terminal half of the molecule contains 6 transmembrane domains, followed by a large cytoplasmic domain with an ATP-binding site, and then a second section with 6 transmembrane domains and an ATP-binding site that shows over 65% of amino acid similarity with the first half of the polypeptide. In 2009, the first structure of a mammalian P-glycoprotein was solved (3G5U). The structure was derived from the mouse MDR3 gene product heterologously expressed in Pichia pastoris yeast. The structure of mouse P-gp is similar to structures of the bacterial ABC transporter MsbA (3B5W and 3B5X) that adopt an inward facing conformation that is believed to be important for binding substrate along the inner leaflet of the membrane. Additional structures (3G60 and 3G61) of P-gp were also solved revealing the binding site(s) of two different cyclic peptide substrate/inhibitors. The promiscuous binding pocket of P-gp is lined with aromatic amino acid side chains. Through Molecular Dynamic (MD) simulations, this sequence was proved to have a direct impact in the transporter's structural stability (in the nucleotide-binding domains) and defining a lower boundary for the internal drug-binding pocket.
Mechanism of action
Substrate enters P-gp either from an opening within the inner leaflet of the membrane or from an opening at the cytoplasmic side of the protein. ATP binds at the cytoplasmic side of the protein. Following binding of each, ATP hydrolysis shifts the substrate into a position to be excreted from the cell. Release of the phosphate (from the original ATP molecule) occurs concurrently with substrate excretion. ADP is released, and a new molecule of ATP binds to the secondary ATP-binding site. Hydrolysis and release of ADP and a phosphate molecule resets the protein, so that the process can start again...
Tissue distribution
P-gp is expressed primarily in certain cell types in the liver, pancreas, kidney, colon, and jejunum. P-gp is also found in brain capillary endothelial cells.
Exogenous inhibitors
Some common pharmacological inhibitors of P-glycoprotein are amiodarone , clarithromycin , ciclosporin , colchicine , diltiazem, erythromycin , felodipine , lansoprazole , omeprazole and other proton pump inhibitors, nifedipine , paroxetine , sertraline, quinidine, tamoxifen and verapamil .
Detecting the activity of the transporter
Radioactive verapamil can be used for measuring P-gp function with positron emission tomography.
P-gp is also used to differentiate transitional B-cells from naive B-cells. Dyes such as Rhodamine123 and MitoTracker Dyes from Invitrogen can be used to make this differentiation.
History
P-gp was first cloned and characterized in 1976. It was shown to be responsible for conferring multidrug resistance upon mutant cultured cancer cells that had developed resistance to cytotoxic drugs.
The structure of P-gp was resolved by x-ray crystallography in 2009.