
EC number 1.18.6.1 ExPASy NiceZyme view | CAS number 9013-04-1 | |
 | ||
Nitrogenases are enzymes (EC 1.18.6.1EC 1.19.6.1) that are produced by certain bacteria, such as cyanobacteria (blue-green algae). These enzymes are responsible for the reduction of nitrogen (N2) to ammonia (NH3). Nitrogenases are the only family of enzymes known to catalyze this reaction, which is a key step in the process of nitrogen fixation. Nitrogen fixation is required for all forms of life, with nitrogen being essential for the biosynthesis of molecules (nucleotides, amino acids) that create plants, animals and other organisms.
Contents
- Structure
- Fe protein
- MoFe protein
- General Mechanism
- Lowe Thorneley Kinetic Model
- Intermediates E0 through E4
- Distal and Alternating Pathways for N2 Fixation
- Other Mechanistic Details
- Nonspecific reactions
- Organisms that synthesize nitrogenase
- Similarity to other proteins
- Measurement of nitrogenase activity
- References
Structure
The equilibrium formation of ammonia from molecular hydrogen and nitrogen has an overall negative enthalpy of reaction (
The nitrogenase complex consists of two proteins:
Fe protein
The Fe protein is a dimer of identical subunits which contains one [Fe4S4] cluster and weighs approximately 60-64kDa. The function of the Fe protein is to transfer electrons from a reducing agent, such as ferredoxin or flavodoxin to the MoFe protein. The transfer of electrons requires an input of chemical energy which comes from the binding and hydrolysis of ATP. The hydrolysis of ATP also causes a conformational change within the nitrogenase complex, bringing the Fe protein and MoFe protein closer together for easier electron transfer.
MoFe protein
The MoFe protein is a heterotetramer consisting of two α subunits and two β subunits, weighing approximately 240-250kDa. The MoFe protein also contains two iron-sulfur clusters, known as P-clusters, located at the interface between the α and β subunits and two FeMo cofactors, within the α subunits.
Electrons from the Fe protein enter the MoFe protein at the P-clusters, which then transfer the electrons to the FeMo cofactors. Each FeMo cofactor then acts as a site for nitrogen fixation, with N2 binding in the central cavity of the cofactor.
General Mechanism
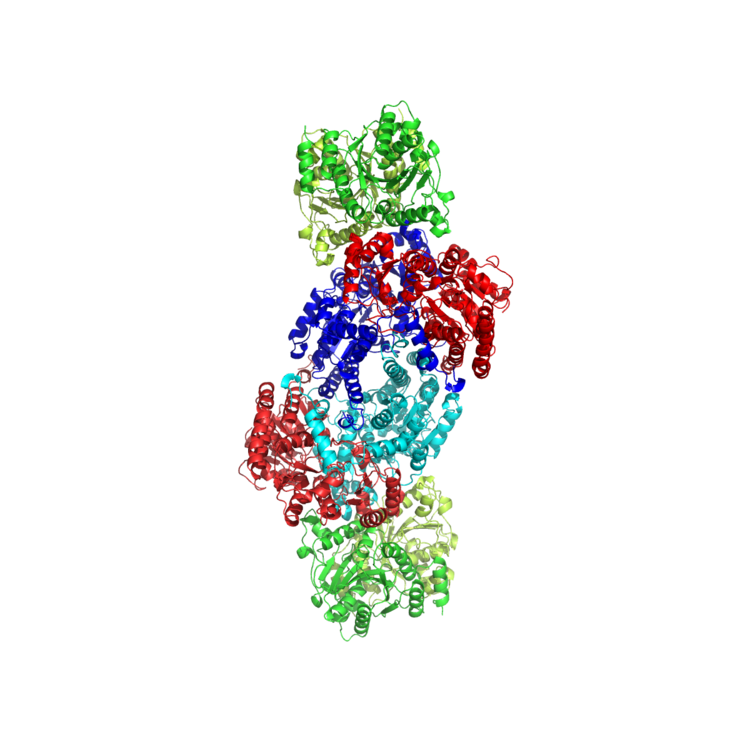
Nitrogenase is an enzyme responsible for catalyzing nitrogen fixation, which is the reduction of nitrogen (N2) to ammonia (NH3) and a process vital to sustaining life on Earth. There are three types of nitrogenase found in various nitrogen-fixing bacteria: molybdenum (Mo) nitrogenase, vanadium (V) nitrogenase, and iron-only (Fe) nitrogenase. Molybdenum nitrogenase, which can be found in diazotrophs and legumes, is the nitrogenase that has been studied the most extensively and thus is the most well characterized. Figures 1-2 display the crystal structure and key catalytic components of molybdenum nitrogenase extracted from Azotobacter vinelandii. Vanadium nitrogenase and iron-only nitrogenase can both be found in select species of Azotobacter as an alternative nitrogenase. Equations 1 and 2 show the balanced reactions of nitrogen fixation in molybdenum nitrogenase and vanadium nitrogenase respectively.
N2 + 8 H+ + 8 e− + 16 MgATP → 2 NH3 + H2 + 16 MgADP + 16 Pi (1)
N2 + 12 H+ + 12 e− + 24 MgATP → 2 NH3 + 3 H2 + 24 MgADP + 24 Pi (2)
All nitrogenases are two-component systems made up of Component I (also known as dinitrogenase) and Component II (also known as dinitrogenase reductase). Component I is a MoFe protein in molybdenum nitrogenase, a VFe protein in vanadium nitrogenase, and a Fe protein in iron-only nitrogenase. Component II is a Fe protein that contains the Fe-S cluster (Figure 3: top), which transfers electrons to Component I. Component I contains 2 key metal clusters: the P-cluster (Figure 3: middle), and the FeMo-cofactor (FeMo-co) (Figure 3: bottom). Mo is replaced by V or Fe in vanadium nitrogenase and iron-only nitrogenase respectively. During catalysis, electrons flow from a pair of ATP molecules within Component II to the Fe-S cluster, to the P-cluster, and finally to the FeMo-co, where reduction of N2 to NH3 takes place.
Lowe-Thorneley Kinetic Model
The reduction of nitrogen to two molecules of ammonia is carried out at the FeMo-co of Component I after the sequential addition of proton and electron equivalents from Component II. Steady state, freeze quench, and stopped-flow kinetics measurements carried out in the 70’s and 80’s by Lowe, Thorneley, and others provided a kinetic basis for this process. The Lowe-Thorneley (LT) kinetic model (depicted in Figure 4) was developed from these experiments and documents the eight correlated proton and electron transfers required throughout the reaction. Each intermediate stage is depicted as En where n = 0-8, corresponding to the number of equivalents transferred. The transfer of four equivalents are required before the productive addition of N2, although reaction of E3 with N2 is also possible. Notably, nitrogen reduction has been shown to require 8 equivalents of protons and electrons as opposed to the 6 equivalents predicted by the balanced chemical reaction.
Intermediates E0 through E4
Spectroscopic characterization of these intermediates has allowed for greater understanding of nitrogen reduction by nitrogenase, however, the mechanism remains an active area of research and debate. Briefly listed below are spectroscopic experiments for the intermediates before the addition of nitrogen:
E0 – This is the resting state of the enzyme before catalysis begins. EPR characterization shows that this species has a spin of 3/2.
E1 – The one electron reduced intermediate has been trapped during turnover under N2. Mӧssbauer spectroscopy of the trapped intermediate indicates that the FeMo-co is integer spin greater than 1.
E2 – This intermediate is proposed to contain the metal cluster in its resting oxidation state with the two added electrons stored in a bridging hydride and the additional proton bonded to a sulfur atom. Isolation of this intermediate in mutated enzymes shows that the FeMo-co is high spin and has a spin of 3/2.
E3 – This intermediate is proposed to be the singly reduced FeMo-co with one bridging hydride and one proton.
E4 – Termed the Janus intermediate after the Roman god of transitions, this intermediate is positioned after exactly half of the electron proton transfers and can either decay back to E0 or proceed with nitrogen binding and finish the catalytic cycle. This intermediate is proposed to contain the FeMo-co in its resting oxidation state with two bridging hydrides and two sulfur bonded protons. This intermediate was first observed using freeze quench techniques with a mutated protein in which residue 70, a valine amino acid, is replaced with isoleucine. This modification prevents substrate access to the FeMo-co. EPR characterization of this isolated intermediate shows a new species with a spin of ½. ENDOR experiments have provided insight into the structure of this intermediate, revealing the presence of two bridging hydrides. 95Mo and 57Fe ENDOR show that the hydrides bridge between two iron centers. Cryoannealing of the trapped intermediate at -20 °C results in the successive loss of two hydrogen equivalents upon relaxation, proving that the isolated intermediate is consistent with the E4 state. The decay of E4 to E2 + H2 and finally to E0 and 2H2 has confirmed the EPR signal associated with the E2 intermediate.
The above intermediates suggest that the metal cluster is cycled between its original oxidation state and a singly reduced state with additional electrons being stored in hydrides. It has alternatively been proposed that each step involves the formation of a hydride and that the metal cluster actually cycles between the original oxidation state and a singly oxidized state.
Distal and Alternating Pathways for N2 Fixation
While the mechanism for nitrogen fixation prior to the Janus E4 complex is generally agreed upon, there are currently two theories for the exact pathway in the second half of the mechanism: the "distal" and the "alternating" pathway (see Figure 5). In the distal pathway, the terminal nitrogen is hydrogenated first, releases ammonia, then the nitrogen directly bound to the metal is hydrogenated. In the alternating pathway, one hydrogen is added to the terminal nitrogen, then one hydrogen is added to the nitrogen directly bound to the metal. This alternating pattern continues until ammonia is released. Because each pathway favors a unique set of intermediates, attempts to determine which path is correct have generally focused on the isolation of said intermediates, such as the nitrido in the distal pathway, and the diazene and hydrazine in the alternating pathway. Attempts to isolate the intermediates in nitrogenase itself have so far been unsuccessful, but the use of model complexes has allowed for the isolation of intermediates that support both sides depending on the metal center used. Studies with Mo generally point towards a distal pathway, while studies with Fe generally point towards an alternating pathway.
Specific support for the distal pathway has mainly stemmed from the work of Schrock and Chatt, who successfully isolated the nitrido complex using Mo as the metal center in a model complex. Specific support for the alternating pathway stems from a few studies. Iron only model clusters have been shown to catalytically reduce N2. Small tungsten clusters have also been shown to follow an alternating pathway for nitrogen fixation. The vanadium nitrogenase releases hydrazine, an intermediate specific to the alternating mechanism. However, the lack of characterized intermediates in the native enzyme itself means that neither pathway has been definitively proven. Furthermore, computational studies have been found to support both sides, depending on whether the reaction site is assumed to be at Mo (distal) or at Fe (alternating) in the MoFe cofactor.
Binding of MgATP is one of the central events to occur in the mechanism employed by nitrogenase. Hydrolysis of the terminal phosphate group of MgATP provides the energy needed to transfer electrons from the Fe protein to the MoFe protein. The binding interactions between the MgATP phosphate groups and the amino acid residues of the Fe protein are well understood by comparing to similar enzymes, while the interactions with the rest of the molecule are more elusive due to the lack of a Fe protein crystal structure with MgATP bound (as of 1996). Three protein residues have been shown to have significant interactions with the phosphates, shown in Figure 6. In the absence of MgATP, a salt bridge exists between residue 15, lysine, and residue 125, aspartic acid. Upon binding, this salt bridge is interrupted. Site-specific mutagenesis has demonstrated that when the lysine is substituted for a glutamine, the protein’s affinity for MgATP is greatly reduced and when the lysine is substituted for an arginine, MgATP cannot bind due to the salt bridge being too strong. The necessity of specifically aspartic acid at site 125 has been shown through noting altered reactivity upon mutation of this residue to glutamic acid. The third residue that has been shown to be key for MgATP binding is residue 16, serine. Site-specific mutagenesis was used to demonstrate this fact. This has led to a model in which the serine remains coordinated to the Mg2+ ion after phosphate hydrolysis in order to facilitate its association with a different phosphate of the now ADP molecule. MgATP binding also induces significant conformational changes within the Fe protein. Site-directed mutagenesis was employed to create mutants in which MgATP binds but does not induce a conformational change. Comparing X-ray scattering data in the mutants versus in the wild-type protein led to the conclusion that the entire protein contracts upon MgATP binding, with a decrease in radius of approximately 2.0 Å.
Other Mechanistic Details
Many mechanistic aspects of catalysis remain unknown. No crystallographic analysis has been reported on substrate bound to nitrogenase. One problem is that the enzyme is generally obtained as a mixture of states. Nitrogenase is able to reduce acetylene, but is inhibited by carbon monoxide, which binds to the enzyme and thereby prevents binding of dinitrogen. Dinitrogen prevent acetylene binding, but acetylene does not inhibit binding of dinitrogen and requires only one electron for reduction to ethylene. Due to the oxidative properties of oxygen, most nitrogenases are irreversibly inhibited by dioxygen, which degradatively oxidizes the Fe-S cofactors. This requires mechanisms for nitrogen fixers to protect nitrogenase from oxygen in vivo. Despite this problem, many use oxygen as a terminal electron acceptor for respiration. Although the ability of some nitrogen fixers such as Azotobacteraceae to employ an oxygen-labile nitrogenase under aerobic conditions has been attributed to a high metabolic rate, allowing oxygen reduction at the cell membrane, the effectiveness of such a mechanism has been questioned at oxygen concentrations above 70 µM (ambient concentration is 230 µM O2), as well as during additional nutrient limitations.
Nonspecific reactions
In addition to dinitrogen reduction, nitrogenases also reduce protons to dihydrogen, meaning nitrogenase is also a dehydrogenase. A list of other reactions carried out by nitrogenases is shown below:
HC≡CH → H2C=CH2N–=N+=O → N2 + H2ON=N=N– → N2 + NH3C≡N−→ CH4, NH3, H3C–CH3, H2C=CH2 (CH3NH2)N≡C–R → RCH3 + NH3C≡N–R → CH4, H3C–CH3, H2C=CH2, C3H8, C3H6, RNH2O=C=S → CO + H2S O=C=O → CO + H2O S=C=N– → H2S + HCN O=C=N– → H2O + HCN, CO + NH3
Furthermore, dihydrogen functions as a competitive inhibitor, carbon monoxide functions as a non-competitive inhibitor, and carbon disulfide functions as a rapid-equilibrium inhibitor of nitrogenase.
Vanadium nitrogenases have also been shown to catalzye the conversion of CO into alkanes through a reaction comparable to Fischer-Tropsch synthesis.
Organisms that synthesize nitrogenase
There are two types of bacteria that synthesize nitrogenase and are required for nitrogen fixation. These are:
Similarity to other proteins
The three subunits of nitrogenase exhibit significant sequence similarity to three subunits of the light-independent version of protochlorophyllide reductase that performs the conversion of protochlorophyllide to chlorophyll. This protein is present in gymnosperms, algae, and photosynthetic bacteria but has been lost by angiosperms during evolution.
Separately, two of the nitrogenase subunits (NifD and NifH) have homologues in methanogens that do not fix nitrogen e.g. Methanocaldococcus jannaschii. Little is understood about the function of these "class IV" nif genes, though they occur in many methanogens. In M. jannaschii they are known to interact with each other and are constitutively expressed.
Measurement of nitrogenase activity
As with many assays for enzyme activity, it is possible to estimate nitrogenase activity by measuring the rate of conversion of the substrate (N2) to the product (NH3). Since NH3 is involved in other reactions in the cell, it is often desirable to label the substrate with 15N to provide accounting or "mass balance" of the added substrate. A more common assay, the acetylene reduction assay or ARA, estimates the activity of nitrogenase by taking advantage of the ability of the enzyme to reduce acetylene gas to ethylene gas. These gases are easily quantified using gas chromatography. Though first used in a laboratory setting to measure nitrogenase activity in extracts of Clostridium pasteurianum cells, ARA has been applied to a wide range of test systems, including field studies where other techniques are difficult to deploy. For example, ARA was used successfully to demonstrate that bacteria associated with rice roots undergo seasonal and diurnal rhythms in nitrogenase activity, which were apparently controlled by the plant.
Unfortunately, the conversion of data from nitrogenase assays to actual moles of N2 reduced (particularly in the case of ARA), is not always straightforward and may either underestimate or overestimate the true rate for a variety of reasons. For example, H2 competes with N2 but not acetylene for nitrogenase (leading to overestimates of nitrogenase by ARA). Bottle or chamber-based assays may produce negative impacts on microbial systems as a result of containment or disruption of the microenvironment through handling, leading to underestimation of nitrogenase. Despite these weaknesses, such assays are very useful in assessing relative rates or temporal patterns in nitrogenase activity.