
 | ||
In chemistry, molecular orbital (MO) theory is a method for determining molecular structure in which electrons are not assigned to individual bonds between atoms, but are treated as moving under the influence of the nuclei in the whole molecule. The spatial and energetic properties of electrons within atoms are fixed by quantum mechanics to form orbitals that contain these electrons. While atomic orbitals contain electrons ascribed to a single atom, molecular orbitals, which surround a number of atoms in a molecule, contain valence electrons between atoms. Molecular orbital theory, which was proposed in the early twentieth century, revolutionized the study of bonding by approximating the positions of bonded electrons—the molecular orbitals—as linear combinations of atomic orbitals (LCAO). These approximations are now made by applying the density functional theory (DFT) or Hartree–Fock (HF) models to the Schrödinger equation.
Contents
Quantitative applications
In this theory, each molecule has a set of molecular orbitals, in which it is assumed that the molecular orbital wave function ψj can be written as a simple weighted sum of the n constituent atomic orbitals χi, according to the following equation:
One may determine cij coefficients numerically by substituting this equation into the Schrödinger's equation and applying the variational principle. The variational principle is a mathematical technique used in quantum mechanics to build up the coefficients of each atomic orbital basis. A larger coefficient means that the orbital basis is composed more of that particular contributing atomic orbital—hence, the molecular orbital is best characterized by that type. This method of quantifying orbital contribution as Linear Combinations of Atomic Orbitals is used in computational chemistry. An additional unitary transformation can be applied on the system to accelerate the convergence in some computational schemes. Molecular orbital theory was seen as a competitor to valence bond theory in the 1930s, before it was realized that the two methods are closely related and that when extended they become equivalent.
History
Molecular orbital theory was developed, in the years after valence bond theory had been established (1927), primarily through the efforts of Friedrich Hund, Robert Mulliken, John C. Slater, and John Lennard-Jones. MO theory was originally called the Hund-Mulliken theory. According to German physicist and physical chemist Erich Hückel, the first quantitative use of molecular orbital theory was the 1929 paper of Lennard-Jones. This paper notably predicted a triplet ground state for the dioxygen molecule which explained its paramagnetism (see Molecular orbital diagram#Dioxygen) before valence bond theory, which came up with its own explanation in 1931. The word orbital was introduced by Mulliken in 1932. By 1933, the molecular orbital theory had been accepted as a valid and useful theory.
Erich Hückel applied molecular orbital theory to unsaturated hydrocarbon molecules starting in 1931 with his Hückel molecular orbital (HMO) method for the determination of MO energies for pi electrons, which he applied to conjugated and aromatic hydrocarbons. This method provided an explanation of the stability of molecules with six pi-electrons such as benzene.
The first accurate calculation of a molecular orbital wavefunction was that made by Charles Coulson in 1938 on the hydrogen molecule. By 1950, molecular orbitals were completely defined as eigenfunctions (wave functions) of the self-consistent field Hamiltonian and it was at this point that molecular orbital theory became fully rigorous and consistent. This rigorous approach is known as the Hartree–Fock method for molecules although it had its origins in calculations on atoms. In calculations on molecules, the molecular orbitals are expanded in terms of an atomic orbital basis set, leading to the Roothaan equations. This led to the development of many ab initio quantum chemistry methods. In parallel, molecular orbital theory was applied in a more approximate manner using some empirically derived parameters in methods now known as semi-empirical quantum chemistry methods.
The success of Molecular Orbital Theory also spawned ligand field theory, which was developed during the 1930s and 1940s as an alternative to crystal field theory.
Types of orbitals
Molecular orbital (MO) theory uses a linear combination of atomic orbitals (LCAO) to represent molecular orbitals resulting from bonds between atoms. These are often divided into bonding orbitals, anti-bonding orbitals, and non-bonding orbitals. A bonding orbital concentrates electron density in the region between a given pair of atoms, so that its electron density will tend to attract each of the two nuclei toward the other and hold the two atoms together. An anti-bonding orbital concentrates electron density "behind" each nucleus (i.e. on the side of each atom which is farthest from the other atom), and so tends to pull each of the two nuclei away from the other and actually weaken the bond between the two nuclei. Electrons in non-bonding orbitals tend to be associated with atomic orbitals that do not interact positively or negatively with one another, and electrons in these orbitals neither contribute to nor detract from bond strength.
Molecular orbitals are further divided according to the types of atomic orbitals they are formed from. Chemical substances will form bonding interactions if their orbitals become lower in energy when they interact with each other. Different bonding orbitals are distinguished that differ by electron configuration (electron cloud shape) and by energy levels.
The molecular orbitals of a molecule can be illustrated in molecular orbital diagrams.
Overview
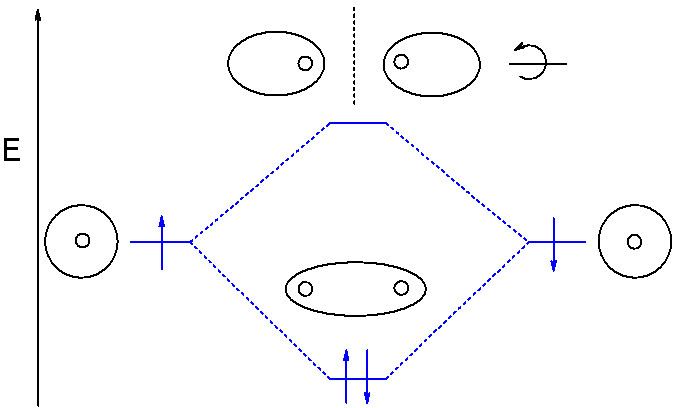
MO theory provides a global, delocalized perspective on chemical bonding. In MO theory, any electron in a molecule may be found anywhere in the molecule, since quantum conditions allow electrons to travel under the influence of an arbitrarily large number of nuclei, as long as they are in eigenstates permitted by certain quantum rules. Thus, when excited with the requisite amount of energy through high-frequency light or other means, electrons can transition to higher-energy molecular orbitals. For instance, in the simple case of a hydrogen diatomic molecule, promotion of a single electron from a bonding orbital to an antibonding orbital can occur under UV radiation. This promotion weakens the bond between the two hydrogen atoms and can lead to photodissociation—the breaking of a chemical bond due to the absorption of light.
Although in MO theory some molecular orbitals may hold electrons that are more localized between specific pairs of molecular atoms, other orbitals may hold electrons that are spread more uniformly over the molecule. Thus, overall, bonding is far more delocalized in MO theory, which makes it more applicable to resonant molecules that have equivalent non-integer bond orders than valence bond (VB) theory. This makes MO theory more useful for the description of extended systems.
An example is the MO description of benzene, C
6H
6, which is an aromatic hexagonal ring of six carbon atoms and three double bonds. In this molecule, 24 of the 30 total valence bonding electrons—24 coming from carbon atoms and 6 coming from hydrogen atoms—are located in 12 σ (sigma) bonding orbitals, which are located mostly between pairs of atoms (C-C or C-H), similarly to the electrons in the valence bond description. However, in benzene the remaining six bonding electrons are located in three π (pi) molecular bonding orbitals that are delocalized around the ring. Two of these electrons are in an MO that has equal orbital contributions from all six atoms. The other four electrons are in orbitals with vertical nodes at right angles to each other. As in the VB theory, all of these six delocalized π electrons reside in a larger space that exists above and below the ring plane. All carbon-carbon bonds in benzene are chemically equivalent. In MO theory this is a direct consequence of the fact that the three molecular π orbitals combine and evenly spread the extra six electrons over six carbon atoms.
In molecules such as methane, CH
4, the eight valence electrons are found in four MOs that are spread out over all five atoms. However, it is possible to transform the MOs into four localized sp3 orbitals. Linus Pauling, in 1931, hybridized the carbon 2s and 2p orbitals so that they pointed directly at the hydrogen 1s basis functions and featured maximal overlap. However, the delocalized MO description is more appropriate for predicting ionization energies and the positions of spectral absorption bands. When methane is ionized, a single electron is taken from the valence MOs, which can come from the s bonding or the triply degenerate p bonding levels, yielding two ionization energies. In comparison, the explanation in VB theory is more complicated. When one electron is removed from an sp3 orbital, resonance is invoked between four valence bond structures, each of which has a single one-electron bond and three two-electron bonds. Triply degenerate T2 and A1 ionized states (CH4+) are produced from different linear combinations of these four structures. The difference in energy between the ionized and ground state gives the two ionization energies.
As in benzene, in substances such as beta carotene, chlorophyll, or heme, some electrons in the π orbitals are spread out in molecular orbitals over long distances in a molecule, resulting in light absorption in lower energies (the visible spectrum), which accounts for the characteristic colours of these substances. This and other spectroscopic data for molecules are well explained in MO theory, with an emphasis on electronic states associated with multicenter orbitals, including mixing of orbitals premised on principles of orbital symmetry matching. The same MO principles also naturally explain some electrical phenomena, such as high electrical conductivity in the planar direction of the hexagonal atomic sheets that exist in graphite. This results from continuous band overlap of half-filled p orbitals and explains electrical conduction. MO theory recognizes that some electrons in the graphite atomic sheets are completely delocalized over arbitrary distances, and reside in very large molecular orbitals that cover an entire graphite sheet, and some electrons are thus as free to move and therefore conduct electricity in the sheet plane, as if they resided in a metal.