
 | ||
Interatomic potentials are mathematical functions for calculating the potential energy of a system of atoms with given positions in space. Interatomic potentials are widely used as the physical basis of molecular mechanics and molecular dynamics simulations in chemistry, molecular physics and materials physics, sometimes in connection with such effects as cohesion, thermal expansion and elastic properties of materials.
Contents
Functional form
Interatomic potentials can be written as a series expansion of functional terms that depend on the position of one, two, three, etc. atoms at a time. Then the total energy of the system V can be written as
Here
Note that in case the pair potential is given per atom pair, in the two-body term the potential should be multiplied by 1/2 as otherwise each bond is counted twice, and similarly the three-body term by 1/6. Alternatively, the summation of the pair term can be restricted to cases
The one-body term is only meaningful if the atoms are in an external field (e.g. an electric field). In the absence of external fields, the potential V should not depend on the absolute position of atoms, but only on the relative positions. This means that the functional form can be rewritten as a function of interatomic distances
In the three-body term
In principle the sums in the expressions run over all N atoms. However, if the range of the interatomic potential is finite, i.e. the potentials
Force calculation
The forces acting between atoms can be obtained by differentiation of the total energy with respect to atom positions. That is, to get the force on atom i one should take the three-dimensional derivative (gradient) with respect to the position of atom i:
For two-body potentials this gradient reduces, thanks to the symmetry with respect to ij in the potential form, to straightforward differentiation with respect to the interatomic distances
Classes of interatomic potentials
Interatomic potentials come in many different varieties, with different physical motivations. Even for single well-known elements such as silicon, a wide variety of potentials quite different in functional form and motivation have been developed. The true interatomic interactions are quantum mechanical in nature, and there is no known way in which the true interactions described by the Schrödinger equation or Dirac equation for all electrons and nuclei could be cast into an analytical functional form. Hence all analytical interatomic potentials are by necessity approximations.
Pair potentials
The arguably simplest widely used interatomic interaction model is the Lennard-Jones potential
where
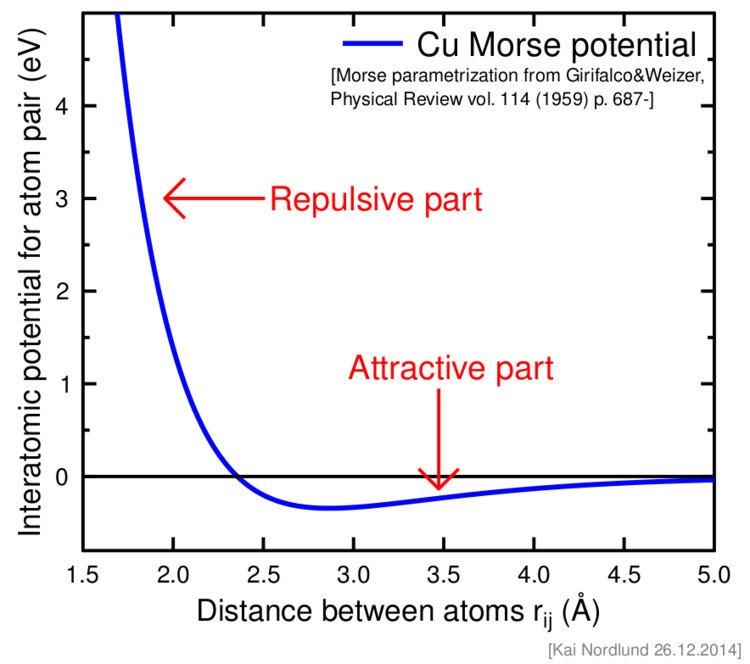
Another simple and widely used pair potential is the Morse potential, which consists simply of a sum of two exponentials.
Here
Ionic materials are often described by a sum of a short-range repulsive term, such as the Buckingham pair potential, and a long-range Coulomb potential giving the ionic interactions between the ions forming the material. The short-range term for ionic materials can also be of many-body character .
Pair potentials have some inherent limitations, like the inability to describe all 3 elastic constants of cubic metals. Hence modern molecular dynamics simulations are to a large extent carried out with different kinds of many-body potentials.
Many-body potentials
The Stilinger-Weber potential is a potential that has a two-body and three-body terms of the standard form
where the three-body term describes how the potential energy changes with bond bending. It was originally developed for pure Si, but has been extended to many other elements and compounds and also formed the basis for other Si potentials.
Metals are very commonly described with what can be called "EAM-like" potentials, i.e. potentials that share the same functional form as the embedded atom model. In these potentials, the total potential energy is written
where
EAM-like potentials are usually implemented as numerical tables. A collection of tables is available at the interatomic potential repository at NIST [1]
Covalently bonded materials are often described by bond order potentials, sometimes also called Tersoff-like or Brenner-like potentials.
These have in general a form that resembles a pair potential:
where the repulsive and attractive part are simple exponential functions similar to those in the Morse potential. However, the strength is modified by the environment of the atom
Repulsive potentials for short-range interactions
For very short interatomic separations, important in radiation material science, the interactions can be described quite accurately with screened Coulomb potentials which have the general form
here φ(r) → 1 when r → 0. Here
Potential fitting
Since the interatomic potentials are approximations, they by necessity all involve parameters that need to be adjusted to some reference values. In simple potentials such as the Lennard-Jones and Morse ones, the parameters can be set directly to match e.g. the equilibrium bond length and bond strength of a dimer molecule or the cohesive energy of a solid . However, many-body potentials often contain tens or even hundreds of adjustable parameters. These can be fit into a larger set of experimental data, or materials properties derived from more fundamental simulation models such as density-functional theory. For solids, a well-constructed many-body potential can often describe at least the equilibrium crystal structure cohesion and lattice constant, linear elastic constants, and basic point defect properties of all the elements and stable compounds well. . The aim of most potential construction and fitting is to make the potential transferable, i.e. that it can describe materials properties that are clearly different from those it was fitted to (for examples of potentials explicitly aiming for this, see e.g.). As an example of demonstrated partial transferability, a review of interatomic potentials of Si found that for instance the Stillinger-Weber and Tersoff III potentials for Si are indeed able to describe several (but certainly not all) materials properties they were not fitted to .
The NIST interatomic potential repository provides a collection of fitted interatomic potentials, either as fitted parameter values or numerical tables of the potential functions.
Reliability of interatomic potentials
Classical interatomic potentials cannot reproduce all phenomena. Sometimes quantum description is necessary. Density functional theory is used to overcome this limitation.