
Specialty oncology ICD-9-CM 202.3 MeSH D031249 | ICD-10 C96.1 DiseasesDB 29792 | |
 | ||
Erdheim–Chester disease (also known as Erdheim–Chester syndrome or polyostotic sclerosing histiocytosis) is a rare disease characterized by the abnormal multiplication of a specific type of white blood cells called histiocytes, or tissue macrophages (technically, this disease is termed a non-Langerhans-cell histiocytosis). Usually, onset is in middle age. The disease involves an infiltration of lipid-laden macrophages, multinucleated giant cells, an inflammatory infiltrate of lymphocytes and histiocytes in the bone marrow, and a generalized sclerosis of the long bones.
Contents
Signs and symptoms
Approximately 500 cases have been reported in the literature to date. ECD affects predominantly adults, with a mean age of 53 years. Long bone involvement is almost universal in ECD patients and is bilateral and symmetrical in nature. More than 50% of cases have some sort of extraskeletal involvement. This can include kidney, skin, brain and lung involvement, and less frequently retroorbital tissue, pituitary gland and heart involvement is observed.
Bone pain is the most frequent of all symptoms associated with ECD and mainly affects the lower limbs, knees and ankles. The pain is often described as mild but permanent, and juxtaarticular in nature. Exophthalmos occurs in some patients and is usually bilateral, symmetric and painless, and in most cases it occurs several years before the final diagnosis. Recurrent pericardial effusion can be a manifestation, as can morphological changes in adrenal size and infiltration.
A review of 59 case studies by Veyssier-Belot, C et al. in 1996 reported the following symptoms in order of frequency of occurrence:
Diagnosis
Radiologic osteosclerosis and histology are the main diagnostic features. Diagnosis can often be difficult because of the rareness of ECD as well as the need to differentiate it from LCH. A diagnosis from neurological imaging may not be definitive. The presence of symmetrical cerebellar and pontine signal changes on T2-weighted images seem to be typical of ECD, however, multiple sclerosis and metabolic diseases must also be considered in the differential diagnosis. ECD is not a common cause of exophthalmos but can be diagnosed by biopsy. However, like all biopsies, this may be inconclusive. Video-assisted thoracoscopic surgery may be used for diagnostic confirmation and also for therapeutic relief of recurrent pericardial fluid drainage.
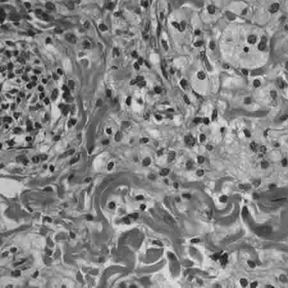
Histology
Histologically, ECD differs from Langerhans cell histiocytosis (LCH) in a number of ways. Unlike LCH, ECD does not stain positive for S-100 proteins or Group 1 CD1a glycoproteins, and electron microscopy of cell cytoplasm does not disclose Birbeck granules. Tissue samples show xanthomatous or xanthogranulomatous infiltration by lipid-laden or foamy histiocytes, and are usually surrounded by fibrosis. Bone biopsy is said to offer the greatest likelihood of reaching a diagnosis. In some, there is histiocyte proliferation, and on staining, the section is CD68+ and CD1a-.
Treatment
Current treatment options include:
All current treatments have had varying degrees of success.
The vinca alkaloids and anthracyclines have been used most commonly in ECD treatment.
Prognosis
Erdheim–Chester disease is associated with high mortality rates. In 2005, the survival rate was below 50% at three years from diagnosis. More recent reports of patients treated with Interferon therapy describe an overall 5-year survival of 68%. Long term survival is currently even more promising, although this impression is not reflected in the recent literature.
History
The first case of ECD was reported by the American pathologist William Chester in 1930, during his visit of the Austrian pathologist Jakob Erdheim in Vienna.
Support groups
The Erdheim–Chester Disease Global Alliance is a support and advocacy group with the goal of raising awareness of and promoting research into ECD. ECD families and patients are also supported by the Histiocytosis Association, Inc.
Patient registry
ECD patients, families, and caregivers are encouraged to join the NIH Rare Lung Diseases Consortium Contact Registry. This is a privacy protected site that provides up-to-date information for individuals interested in the latest scientific news, trials, and treatments related to rare lung diseases.
Media
In the TV show House Season 2 episode 17, All In, the final diagnosis of a 6-year-old boy who presents with bloody diarrhea and ataxia is Erdheim–Chester disease.