
 | ||
Long-term potentiation (LTP), thought to be the cellular basis for learning and memory, involves a specific signal transmission process that underlies synaptic plasticity. Among the many mechanisms responsible for the maintenance of synaptic plasticity is the cadherin–catenin complex. By forming complexes with intracellular catenin proteins, neural cadherins (N-cadherins) serve as a link between synaptic activity and synaptic plasticity, and play important roles in the processes of learning and memory.
Contents
- Structure of the cadherincatenin complex
- Regulation of adhesion
- Regulation of cadherin surface levels turnover and stability
- Intracellular binding partners
- Signal tranduction pathways
- Role in receptor traffickingstability
- Regulation of gene expression
- References
N-cadherins are believed to be involved in mediating LTP and the synaptic changes underlying learning and memory. During embryonic development, cadherins are initially widely distributed, but they become gradually more localized to pre- and post-synaptic sites while synapses are being formed. Blocking cadherin function with specific proteins does not affect basal synaptic properties, but it can impair the induction of LTP.
Structure of the cadherin–catenin complex
N-cadherins are transmembrane proteins expressed in the majority of CNS synapses. N-cadherins are most commonly expressed on both the presynaptic active zone and postsynaptic density (PSD) regions, and contain both extracellular Ca2+ binding domains as well as intracellular domains for binding their protein partners. A common binding partner for Cadherins are intracellular catenin proteins, specifically the three different subtypes, α-catenins, β-catenins, and p120ctn family catenins. β-catenins and p120ctns bind cadherin’s intracellular domain at the distal and proximal regions respectively. α-catenins, when in monomeric form, will associate with the cadherin-catenin complex via β-catenins. In homodimeric form, α-catenins do not bind β- catenins, but preferentially bind F-actin and other proteins promoting F-actin polymerization. Each catenin subtype and its interaction with cadherins plays a distinct role in the mediation of synaptic plasticity and spine structure.
Regulation of adhesion
Evidence suggest that N-Cadherins stabilize the connection between the presynaptic terminal and postsynaptic spine and that this stabilization increases the likelihood that released glutamate will bind receptors on the postsynaptic neuron. At basal levels of synaptic activity, N-cadherins are largely monomers and are thus weakly adhesive to cadherins located in the cell on the opposite side of the synapse. The influx of Ca2+ through NMDARs, promotes the dimerization of N-cadherins. Dimerized cadherins readily bind to their presynaptic cadherin partners. Inhibition of N-cadherin binding via blocking antibodies prevents the induction of late phase L-long term potentiation, suggesting that the adhesive property of dimeric N-cadherin is necessary for late phase L-LTP. Additionally, KCl depolarization of the presynaptic axon both confers protease resistance to N-cadherins and disperses them throughout the PSD from their original clustering in synaptic puncta, thus increasing their efficacy for cell adhesion.
N-cadherin adhesion further stabilizes the synapse by enabling AMPAR-activation-induced spine head expansion. This morphological change helps to prevent further synaptic modifications that could jeopardize the information held by the already existing synaptic connections. Spine head expansion accomplishes this by reducing the NMDAR to AMPAR ratio, creating a proportionally smaller calcium influx, as well as by allowing for faster calcium diffusion out of the spine. The rapid removal of calcium prevents it from initiating the post-translational modifications that would further alter synaptic strength. Overexpression of an N-cadherin mutant incapable of adhesion prevents spine head expansion, demonstrating N-cadherin’s essential role in this process.
Regulation of cadherin surface levels, turnover, and stability
Regulation mechanisms differ in their synthesis requirements and their temporal initiation. One mode of cadherin regulation is surface stabilization, a fairly rapid process (occurring approximately 100 minutes after activity) that is independent of protein synthesis.
NMDAR activity reduces phosphorylation of β-catenin at tyr-654, thereby inhibiting N-cadherin endocytosis and facilitating surface retention and expression.
Surface expression is also regulated via protocadherin-mediated adhesion. Protocadherin-alpha and protocadherin-gamma interact to form a protein complex that enhances the surface expression of each cadherin subtype.
Cadherin expression is also regulated by activity-induced internalization, which occurs much later than surface stabilization (an average of 4 hours after the stimulus). Internalization is dependent on protein synthesis, and p120 catenin proteins (p120ctn) are implicated in the turnover, degradation and ‘clustering’ of cadherins into the adhesive junctions at the synapse. P120 ctn proteins are thought to either inhibit endocytosis of neural cadherins, or act at the cell surface to control cadherin turnover. Down-regulation of p120 ctn leads to greater cadherin endocytosis and prevents adhesion between the pre and post-synaptic neuron. Such evidence strongly suggests that p120 is necessary for cadherin stability.
Intracellular binding partners
β-catenin localizes reserve pool vesicles (RPVs) at presynaptic sites. The deletion of β-catenin in vivo results in a decrease in the number of RPVs localized in the synaptic site and an increase in RPVs dispersed along the axon. Moreover, RPVs were unresponsive to continuous trains of stimulation of the presynaptic neuron in B-catenin knockouts. This effect of B-catenin is independent of cadherin-mediated adhesion, and is instead modulated by PDZ binding proteins such as Veli to cadherin. Additionally, BDNF/TrkB signaling leads to the phosphorylation of β-catenin at its Y654 site, causing the β-catenin–cadherin complex to dissolve and consequently increasing synaptic vesicle mobility. Catenins also bind many scaffolding proteins, receptors, kinases and phosphatases. For example, the cadherin-α-catenin complex binds the actin cytoskeleton, though whether it binds via binding proteins or direct interactions is unknown.
Signal tranduction pathways
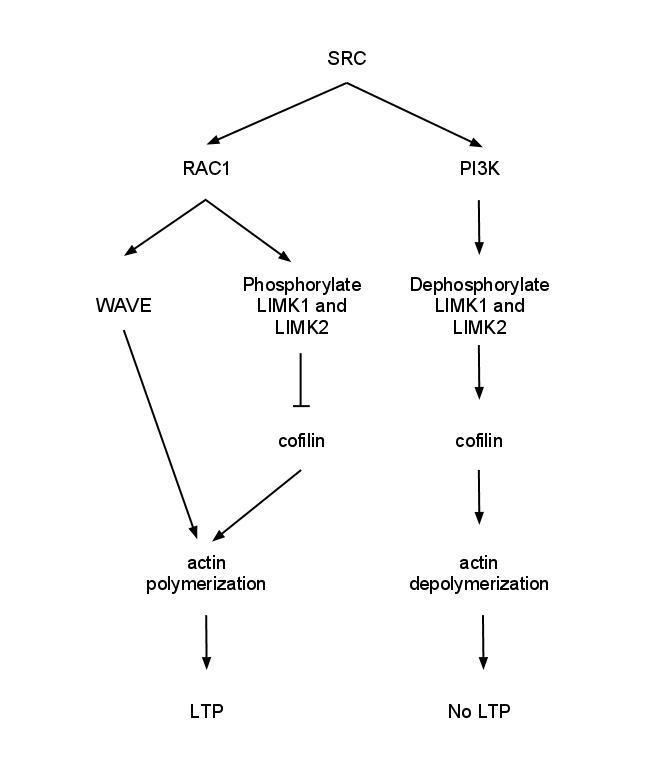
p120ctn can signal through guanine nucleotide exchange factors (GEFs) and GAPs to activate the Rho family of GTPases. RhoA must be inhibited by P120ctn to maintain spine density and length. Rac (GTPase) inhibition remediates a reduction of spine density in p120ctn deficiency. E-cadherin binding to p120ctn may activate SRC leading to activation of Rac1 which results in phosphorylation of LIMK1 and LIMK2 to deactivate cofilin causing G-actin polymerization. Meanwhile, phosphorylation of PI3K by SRC activates RhoA which leads to activation of cofilin-P and disruption of filamentous actin. When the WAVE-1 gene was disurpted in mice, it resulted in cognitive defects such as losses in learning and memory implicating the WAVE-1 branch of the Rac pathway. Using an in vivo dentate gyrus LTP model, it was shown that LTP induction is associated with an increase in F-actin in the dendritic spines, and this is a long lasting change. It was shown that NMDA receptor activation is required for this effect. Furthermore, the use of latrunculin A was able to impair late phase LTP in this model, again suggesting that actin remodelling is necessary for LTP.
Role in receptor trafficking/stability
When the glutamate receptor binding protein ABP and p120ctn are co-expressed, anti-p120ctn serum pulls out a complex containing both proteins from cell lysates. The same occurs for a protein similar to ABP, called GRIP. cDNA screening and yeast mating assays demonstrate that the PDZ domain-binding motif at the p120ctn C-terminus enables such interactions. Co-IP data shows that p120ctn can simultaneously complex with cadherin and either ABP or GRIP. Dominant negative p120ctn fragments, which failed to interact with ABP and GRIP, impaired the stabilization of GluR2 and GluR3 AMPAR subunits, respectively, at the plasma membrane. Co-localization of p120ctn with PSD-95 suggests that cadherin-p120ctn-ABP/GRIP complexes anchor AMPARs at the postsynaptic density, but it is unclear whether anchoring also occurs at perisynaptic sites.
Regulation of gene expression
Cadherins and catenins have been shown to be involved in regulating gene expression, an important process in synaptic plasticity. Glutamate binding to NMDA upregulates the production of N-cadherin’s intracellular domain peptide, N-cad/CTF2, an effect blocked by the NMDA receptor antagonist, APV. Transfection of N-cad/CTF2 decreases nuclear CBP (CREB-binding protein) and increases cytosolic CBP. Furthermore, N-cad/CTF2 co-immunoprecipitates with CBP, and transfection of N-cad/CTF2 reduces CBP steady state levels, consequently impairing CREB-containing DNA complex formation (See figures on the right).
Expression of lymphoid enhancer-binding factor 1 (LEF-1) triggers the translocation of β-catenin to the nucleus, where it upregulates transcription, and transfection of N-cadherin or α-catenin reverses this effect. Additionally, treating neurons with NMDAR agonist causes cleavage of the β-catenin N-terminus, and the C-terminal fragments translocate to the nucleus, where, as transfection experiments show, β-catenin increases T-cell factor (TCF) dependent transcription.