
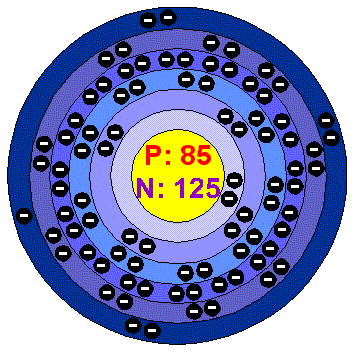
Name, symbol astatine, At Symbol At Atomic number 85 Melting point 301.8 °C Discovered 1940 | Electron configuration [Xe] 4f145d106s26p5 Atomic mass 210 u Boiling point 336.8 °C | |
 | ||
Pronunciation /ˈæstətiːn/ or /ˈæstətɪn/AS-tə-teen or AS-tə-tin Appearance unknown, probably metallic | ||
Sean astatine element project
Astatine is a radioactive chemical element with chemical symbol At and atomic number 85, and is the rarest naturally occurring element on the Earth's crust. It occurs on Earth as the decay product of various heavier elements. All its isotopes are short-lived; the most stable is astatine-210, with a half-life of 8.1 hours. Elemental astatine has never been viewed because any macroscopic sample would be immediately vaporized by its radioactive heating. It has yet to be determined if this obstacle could be overcome with sufficient cooling.
Contents
- Sean astatine element project
- Astatine periodic table of videos
- Characteristics
- Physical
- Chemical
- Compounds
- History
- Isotopes
- Natural occurrence
- Formation
- Separation methods
- Dry
- Wet
- Uses and precautions
- References

The bulk properties of astatine are not known with any certainty. Many of these have been estimated based on its periodic table position as a heavier analog of iodine, and a member of the halogens – the group of elements including fluorine, chlorine, bromine, and iodine. It is likely to have a dark or lustrous appearance and may be a semiconductor or possibly a metal; it probably has a higher melting point than that of iodine. Chemically, several anionic species of astatine are known and most of its compounds resemble those of iodine. It also shows some metallic behavior, including being able to form a stable monatomic cation in aqueous solution (unlike the lighter halogens).

Dale R. Corson, Kenneth Ross MacKenzie, and Emilio G. Segrè synthesized the element at the University of California, Berkeley in 1940, naming it after the Greek astatos (ἄστατος), "unstable". Four isotopes of astatine were subsequently found in nature, although it is the least abundant of all the naturally occurring elements, with much less than one gram being present at any given time in the Earth's crust. Neither the most stable isotope astatine-210 nor the medically useful astatine-211 occurs naturally. They can only be produced synthetically, usually by bombarding bismuth-209 with alpha particles.
Astatine periodic table of videos
Characteristics

Astatine is an extremely radioactive element; all its isotopes have short half-lives of 8.1 hours or less, decaying into other astatine isotopes, bismuth, polonium or radon. Most of its isotopes are very unstable with half-lives of one second or less. Of the first 101 elements in the periodic table, only francium is less stable.
The bulk properties of astatine are not known with any certainty. Research is limited by its short half-life, which prevents the creation of weighable quantities. A visible piece of astatine would immediately vaporize itself because of the heat generated by its intense radioactivity. It remains to be seen if, with sufficient cooling, a macroscopic quantity of astatine could be deposited as a thin film. Astatine is usually classified as either a nonmetal or a metalloid; metal formation has also been predicted.
Physical
Most of the physical properties of astatine have been estimated (by interpolation or extrapolation), using theoretically or empirically derived methods. For example, halogens get darker with increasing atomic weight – fluorine is nearly colorless, chlorine is yellow-green, bromine is red-brown, and iodine is dark gray/violet. Astatine is sometimes described as probably being a black solid (assuming it follows this trend), or as having a metallic appearance (if it is a metalloid or a metal). The melting and boiling points of astatine are also expected to follow the trend seen in the halogen series, increasing with atomic number. On this basis they are estimated to be 575 and 610 K (302 and 337 °C; 575 and 638 °F), respectively. Some experimental evidence suggests astatine may have lower melting and boiling points than those implied by the halogen trend. Astatine sublimes less readily than does iodine, having a lower vapor pressure. Even so, half of a given quantity of astatine will vaporize in approximately an hour if put on a clean glass surface at room temperature. The absorption spectrum of astatine in the middle ultraviolet region has lines at 224.401 and 216.225 nm, suggestive of 6p to 7s transitions.
The structure of solid astatine is unknown. As an analogue of iodine it may have an orthorhombic crystalline structure composed of diatomic astatine molecules, and be a semiconductor (with a band gap of 0.7 eV). Alternatively, if condensed astatine forms a metallic phase, as has been predicted, it may have a monatomic face-centered cubic structure. Evidence for (or against) the existence of diatomic astatine (At2) is sparse and inconclusive. Some sources state that it does not exist, or at least has never been observed, while other sources assert or imply its existence. Despite this controversy, many properties of diatomic astatine have been predicted; for example, its bond length would be 300 ±10 pm, dissociation energy 83.7 ±12.5 kJ·mol−1, and heat of vaporization (∆Hvap) 54.39 kJ·mol−1. The latter figure means that astatine may (at least) be metallic in the liquid state on the basis that elements with a heat of vaporization greater than ~42 kJ·mol−1 are metallic when liquid; diatomic iodine, with a value of 41.71 kJ·mol−1, falls just short of the threshold figure.
Chemical
The chemistry of astatine is "clouded by the extremely low concentrations at which astatine experiments have been conducted, and the possibility of reactions with impurities, walls and filters, or radioactivity by-products, and other unwanted nano-scale interactions." Many of its apparent chemical properties have been observed using tracer studies on extremely dilute astatine solutions, typically less than 10−10 mol·L−1. Some properties – such as anion formation – align with other halogens. Astatine has some metallic characteristics as well, such as plating onto a cathode, coprecipitating with metal sulfides in hydrochloric acid, and forming a stable monatomic cation in aqueous solution. It forms complexes with EDTA, a metal chelating agent, and is capable of acting as a metal in antibody radiolabeling; in some respects astatine in the +1 state is akin to silver in the same state. Most of the organic chemistry of astatine is, however, analogous to that of iodine.
Astatine has an electronegativity of 2.2 on the revised Pauling scale – lower than that of iodine (2.66) and the same as hydrogen. In hydrogen astatide (HAt) the negative charge is predicted to be on the hydrogen atom, implying that this compound should instead be referred to as astatine hydride. That would be consistent with the electronegativity of astatine on the Allred–Rochow scale (1.9) being less than that of hydrogen (2.2). The electron affinity of astatine is predicted to be reduced by one-third because of spin-orbit interactions.
Compounds
Less reactive than iodine, astatine is the least reactive of the halogens, although its compounds have been synthesized in microscopic amounts and studied as intensively as possible before their radioactive disintegration. The reactions involved have been typically tested with dilute solutions of astatine mixed with larger amounts of iodine. Acting as a carrier, the iodine ensures there is sufficient material for laboratory techniques (such as filtration and precipitation) to work. Like iodine, astatine has been shown to adopt odd-numbered oxidation states ranging from −1 to +7.
Only a few compounds with metals have been reported, in the form of astatides of sodium, palladium, silver, thallium, and lead. Some characteristic properties of silver and sodium astatide, and the other hypothetical alkali and alkaline earth astatides, have been estimated by extrapolation from other metal halides.
The formation of an astatine compound with hydrogen – usually referred to as hydrogen astatide – was noted by the pioneers of astatine chemistry. As mentioned, there are grounds for instead referring to this compound as astatine hydride. It is easily oxidized; acidification by dilute nitric acid gives the At0 or At+ forms, and the subsequent addition of silver(I) may only partially, at best, precipitate astatine as silver(I) astatide (AgAt). Iodine, in contrast, is not oxidized, and precipitates readily as silver(I) iodide.
Astatine is known to bind to boron, carbon, and nitrogen. Various boron cage compounds have been prepared with At–B bonds, these being more stable than At–C bonds. Carbon tetraastatide (CAt4) has been synthesized. Astatine can replace a hydrogen atom in benzene to form astatobenzene C6H5At; this may be oxidized to C6H5AtCl2 by chlorine. By treating this compound with an alkaline solution of hypochlorite, C6H5AtO2 can be produced. In the molecules dipyridine-astatine(I) perchlorate [At(C5H5N)2][ClO4] and the analogous nitrate, the astatine atom is bonded to each nitrogen atom in the two pyridine rings.
With oxygen, there is evidence of the species AtO−, AtO−
2, and AtO+ in aqueous solution, formed by the reaction of astatine with an oxidant such as elemental bromine or (in the last case) by sodium persulfate in a solution of perchloric acid. The well characterized AtO−
3 anion can be obtained by, for example, the oxidation of astatine with potassium hypochlorite in a solution of potassium hydroxide. Preparation of lanthanum triastatinate La(AtO3)3, following the oxidation of astatine by a hot Na2S2O8 solution, has been reported. Further oxidation of AtO−
3, such as by xenon difluoride (in a hot alkaline solution) or periodate (in a neutral or alkaline solution), yields the perastatate ion AtO−
4; this is only stable in neutral or alkaline solutions. Astatine is also thought to be capable of forming cations in salts with oxyanions such as iodate or dichromate; this is based on the observation that, in acidic solutions, monovalent or intermediate positive states of astatine coprecipitate with the insoluble salts of metal cations such as silver(I) iodate or thallium(I) dichromate.
Astatine may form bonds to the other chalcogens; these include S7At+ and At(CSN)−
2 with sulfur, a coordination selenourea compound with selenium, and an astatine–tellurium colloid with tellurium.
Astatine is known to react with its lighter homologs iodine, bromine, and chlorine in the vapor state; these reactions produce diatomic interhalogen compounds with formulas AtI, AtBr, and AtCl. The first two compounds may also be produced in water – astatine reacts with iodine/iodide solution to form AtI, whereas AtBr requires (aside from astatine) an iodine/iodine monobromide/bromide solution. The excess of iodides or bromides may lead to AtBr−
2 and AtI−
2 ions, or in a chloride solution, they may produce species like AtCl−
2 or AtBrCl−
via equilibrium reactions with the chlorides. Oxidation of the element with dichromate (in nitric acid solution) showed that adding chloride turned the astatine into a molecule likely to be either AtCl or AtOCl. Similarly, AtOCl−
2 or AtCl−
2 may be produced. Polyhalides PdAtI2, CsAtI2, TlAtI2, and PbAtI are known or presumed to have been precipitated. In a plasma ion source mass spectrometer, the ions [AtI]+, [AtBr]+, and [AtCl]+ have been formed by introducing lighter halogen vapors into a helium-filled cell containing astatine, supporting the existence of stable neutral molecules in the plasma ion state. No astatine fluorides have been discovered yet. Their absence has been speculatively attributed to the extreme reactivity of such compounds, including the reaction of an initially formed fluoride with the walls of the glass container to form a non-volatile product. Thus, although the synthesis of an astatine fluoride is thought to be possible, it may require a liquid halogen fluoride solvent, as has already been used for the characterization of radon fluoride.
History
In 1869, when Dmitri Mendeleev published his periodic table, the space under iodine was empty; after Niels Bohr established the physical basis of the classification of chemical elements, it was suggested that the fifth halogen belonged there. Before its officially recognized discovery, it was called "eka-iodine" (from Sanskrit eka – "one") to imply it was one space under iodine (in the same manner as eka-silicon, eka-boron, and others). Scientists tried to find it in nature; given its extreme rarity, these attempts resulted in several false discoveries.
The first claimed discovery of eka-iodine was made by Fred Allison and his associates at the Alabama Polytechnic Institute (now Auburn University) in 1931. The discoverers named element 85 "alabamine", and assigned it the symbol Ab, designations that were used for a few years. In 1934, H. G. MacPherson of University of California, Berkeley disproved Allison's method and the validity of his discovery. There was another claim in 1937, by the chemist Rajendralal De. Working in Dacca in British India (now Dhaka in Bangladesh), he chose the name "dakin" for element 85, which he claimed to have isolated as the thorium series equivalent of radium F (polonium-210) in the radium series. The properties he reported for dakin do not correspond to those of astatine; moreover, astatine is not found in the thorium series, and the true identity of dakin is not known.
In 1936, a team of Romanian physicist Horia Hulubei and French physicist Yvette Cauchois claimed to have discovered element 85 via X-ray analysis. In 1939 they published another paper which supported and extended previous data. In 1944, Hulubei published a summary of data he had obtained up to that time, claiming it was supported by the work of other researchers. He chose the name "dor", presumably from the Romanian for "longing" [for peace], as World War II had started five years earlier. As Hulubei was writing in French, a language which does not accommodate the "ine" suffix, dor would likely have been rendered in English as "dorine", had it been adopted. In 1947, Hulubei's claim was effectively rejected by the Austrian chemist Friedrich Paneth, who would later chair the IUPAC committee responsible for recognition of new elements. Even though Hulubei's samples did contain astatine, his means to detect it were too weak, by current standards, to enable correct identification. He had also been involved in an earlier false claim as to the discovery of element 87 (francium) and this is thought to have caused other researchers to downplay his work.
In 1940, the Swiss chemist Walter Minder announced the discovery of element 85 as the beta decay product of radium A (polonium-218), choosing the name "helvetium" (from Helvetia, "Switzerland"). Karlik and Bernert were unsuccessful in reproducing his experiments, and subsequently attributed Minder's results to contamination of his radon stream (radon-222 is the parent isotope of polonium-218). In 1942, Minder, in collaboration with the English scientist Alice Leigh-Smith, announced the discovery of another isotope of element 85, presumed to be the product of thorium A (polonium-216) beta decay. They named this substance "anglo-helvetium", but Karlik and Bernert were again unable to reproduce these results.
Later in 1940, Dale R. Corson, Kenneth Ross MacKenzie, and Emilio Segrè isolated the element at the University of California, Berkeley. Instead of searching for the element in nature, the scientists created it by bombarding bismuth-209 with alpha particles in a cyclotron (particle accelerator) to produce, after emission of two neutrons, astatine-211. The discoverers, however, did not immediately suggest a name for the element; this was because an element created synthetically at the moment was not seen as a completely valid one, created in "invisible quantities" and not yet discovered in nature; in addition, chemists were reluctant to recognize radioactive isotopes as legitimately as stable ones. In 1943, astatine was found as a product of two naturally occurring decay chains by Berta Karlik and Traude Bernert, first in the so-called uranium series, and then in the actinium series. (Since then, astatine has been determined in a third decay chain, the neptunium series.) Friedrich Paneth in 1946 called to finally recognize synthetic elements, quoting, among other reasons, recent confirmation of their natural occurrence, and proposed the discoverers of the newly discovered unnamed elements name these elements. In early 1947, Nature published discoverers' suggestions; a letter from Corson, MacKenzie, and Segrè suggested the name "astatine" coming from the Greek astatos (αστατος) meaning "unstable", because of its propensity for radioactive decay, with the ending "-ine", found in the names of the four previously discovered halogens. The name was also chosen to continue the tradition of the four stable halogens, where the name referred to a property of the element.
Corson and his colleagues classified astatine as a metal on the basis of its analytical chemistry. Subsequent investigators reported iodine-like, cationic, or amphoteric behavior. In a 2003 retrospective, Corson wrote that "some of the properties [of astatine] are similar to iodine … it also exhibits metallic properties, more like its metallic neighbors Po and Bi."
Isotopes
There are 39 known isotopes of astatine, with atomic masses (mass numbers) of 191–229. Theoretical modeling suggests that 37 more isotopes could exist. No stable or long-lived astatine isotope has been observed nor is one expected to exist.
Astatine's alpha decay energies follow the same trend as for other heavy elements. Lighter astatine isotopes have quite high energies of alpha decay, which become lower as the nuclei become heavier. Astatine-211 has a significantly higher energy than the previous isotope, because it has a nucleus with 126 neutrons, and 126 is a magic number corresponding to a filled neutron shell. Despite having a similar half-life to the previous isotope (8.1 hours for astatine-210 and 7.2 hours for astatine-211), the alpha decay probability is much higher for the latter: 41.81% against only 0.18%. The two following isotopes release even more energy, with astatine-213 releasing the most energy. For this reason, it is the shortest-lived astatine isotope. Even though heavier astatine isotopes release less energy, no long-lived astatine isotope exists, because of the increasing role of beta decay (electron emission). This decay mode is especially important for astatine; as early as 1950 it was postulated that all isotopes of the element undergo beta decay. Beta decay modes have been found for all astatine isotopes except astatine-213, -214, -215, and -216m. Astatine-210 and lighter isotopes exhibit beta plus decay (positron emission), astatine-216 and heavier isotopes exhibit beta (minus) decay, and astatine-212 decays via both modes, while astatine-211 undergoes electron capture.
The most stable isotope is astatine-210, which has a half-life of 8.1 hours. The primary decay mode is beta plus, to the relatively long-lived (in comparison to astatine isotopes) alpha emitter polonium-210. In total, only five isotopes have half-lives exceeding one hour (astatine-207 to -211). The least stable ground state isotope is astatine-213, with a half-life of 125 nanoseconds. It undergoes alpha decay to the extremely long-lived bismuth-209.
Astatine has 24 known nuclear isomers, which are nuclei with one or more nucleons (protons or neutrons) in an excited state. A nuclear isomer may also be called a "meta-state", meaning the system has more internal energy than the "ground state" (the state with the lowest possible internal energy), making the former likely to decay into the latter. There may be more than one isomer for each isotope. The most stable of these nuclear isomers is astatine-202m1, which has a half-life of about 3 minutes, longer than those of all the ground states bar those of isotopes 203–211 and 220. The least stable is astatine-214m1; its half-life of 265 nanoseconds is shorter than those of all ground states except that of astatine-213.
Natural occurrence
Astatine is the rarest naturally occurring element. The total amount of astatine in the Earth's crust (quoted mass 2.36 × 1025 grams) is estimated to be less than one gram at any given time.
Any astatine present at the formation of the Earth has long since disappeared; the four naturally occurring isotopes (astatine-215, -217, -218 and -219) are instead continuously produced as a result of the decay of radioactive thorium and uranium ores, and trace quantities of neptunium-237. The landmass of North and South America combined, to a depth of 16 kilometers (10 miles), contains only about one trillion astatine-215 atoms at any given time (around 3.5 × 10−10 grams). Astatine-217 is produced via the radioactive decay of neptunium-237. Primordial remnants of the latter isotope—due to its relatively short half-life of 2.14 million years—are no longer present on Earth. However trace amounts occur naturally as a product of transmutation reactions in uranium ores. Astatine-218 was the first astatine isotope discovered in nature. Astatine-219, with a half-life of 56 seconds, is the longest lived of the naturally occurring isotopes.
Isotopes of astatine are sometimes not listed as naturally occurring because of misconceptions that there are no such isotopes, or discrepancies in the literature. Astatine-216 has been counted as a naturally occurring isotope but reports of its observation (which were described as doubtful) have not been confirmed.
Formation
Astatine was first produced by bombarding bismuth-209 with energetic alpha particles, and this is still the major route used to create the relatively long-lived isotopes astatine-209 through astatine-211. Astatine is only produced in minuscule quantities, with modern techniques allowing production runs of up to 6.6 gigabecquerels (about 86 nanograms or 2.47 × 1014 atoms). Synthesis of greater quantities of astatine using this method is constrained by the limited availability of suitable cyclotrons and the prospect of melting the target. Solvent radiolysis due to the cumulative effect of astatine decay is a related problem. With cryogenic technology, microgram quantities of astatine might be able to be generated via proton irradiation of thorium or uranium to yield radon-211, in turn decaying to astatine-211. Contamination with astatine-210 is expected to be a drawback of this method.
The most important isotope is astatine-211, the only one in commercial use. To produce the bismuth target, the metal is sputtered onto a gold, copper, or aluminium surface at 50 to 100 milligrams per square centimeter. Bismuth oxide can be used instead; this is forcibly fused with a copper plate. The target is kept under a chemically neutral nitrogen atmosphere, and is cooled with water to prevent premature astatine vaporization. In a particle accelerator, such as a cyclotron, alpha particles are collided with the bismuth. Even though only one bismuth isotope is used (bismuth-209), the reaction may occur in three possible ways, producing astatine-209, astatine-210, or astatine-211. In order to eliminate undesired nuclides, the maximum energy of the particle accelerator is set to a value (optimally 29.17 MeV) above that for the reaction producing astatine-211 (to produce the desired isotope) and below the one producing astatine-210 (to avoid producing other astatine isotopes).
Separation methods
Since astatine is the main product of the synthesis, after its formation it must only be separated from the target and any significant contaminants. Several methods are available, "but they generally follow one of two approaches—dry distillation or [wet] acid treatment of the target followed by solvent extraction." The methods summarized below are modern adaptations of older procedures, as reviewed by Kugler and Keller. Pre-1985 techniques more often addressed the elimination of co-produced toxic polonium; this requirement is now mitigated by capping the energy of the cyclotron irradiation beam.
Dry
The astatine-containing cyclotron target is heated to a temperature of around 650 °C. The astatine volatilizes and is condensed in (typically) a cold trap. Higher temperatures of up to around 850 °C may increase the yield, at the risk of bismuth contamination from concurrent volatilization. Redistilling the condensate may be required to minimize the presence of bismuth (as bismuth can interfere with astatine labeling reactions). The astatine is recovered from the trap using one or more low concentration solvents such as sodium hydroxide, methanol or chloroform. Astatine yields of up to around 80% may be achieved. Dry separation is the method most commonly used to produce a chemically useful form of astatine.
Wet
The bismuth (or sometimes bismuth trioxide) target is dissolved in, for example, concentrated nitric or perchloric acid. Astatine is extracted using an organic solvent such as butyl or isopropyl ether, or thiosemicarbazide. A separation yield of 93% using nitric acid has been reported, falling to 72% by the time purification procedures were completed (distillation of nitric acid, purging residual nitrogen oxides, and redissolving bismuth nitrate to enable liquid-liquid extraction). Wet methods involve "multiple radioactivity handling steps" and are not well suited for isolating larger quantities of astatine. They can enable the production of astatine in a specific oxidation state and may have greater applicability in experimental radiochemistry.
Uses and precautions
Newly formed astatine-211 is the subject of ongoing research in nuclear medicine. It must be used quickly as it decays with a half-life of 7.2 hours; this is long enough to permit multistep labeling strategies. Astatine-211 has potential for targeted alpha particle radiotherapy, since it decays either via emission of an alpha particle (to bismuth-207), or via electron capture (to an extremely short-lived nuclide, polonium-211, which undergoes further alpha decay). Polonium X-rays emitted as a result of the electron capture branch, in the range of 77–92 keV, enable the tracking of astatine in animals and patients. Although astatine-210 has a slightly longer half-life, it is wholly unsuitable because it decays to the extremely toxic polonium-210.
The principal medicinal difference between astatine-211 and iodine-131 (a radioactive iodine isotope also used in medicine) is that iodine-131 emits high-energy beta particles, and astatine does not. Beta particles have much greater penetrating power through tissues than do the much heavier alpha particles. An average alpha particle released by astatine-211 can travel up to 70 µm through surrounding tissues; an average-energy beta particle emitted by iodine-131 can travel nearly 30 times as far, to about 2 mm. The short half-life and limited penetrating power of alpha radiation through tissues offers advantages in situations where the "tumor burden is low and/or malignant cell populations are located in close proximity to essential normal tissues." Significant morbidity in cell culture models of human cancers has been achieved with from one to ten astatine-211 atoms bound per cell.
Several obstacles have been encountered in the development of astatine-based radiopharmaceuticals for cancer treatment. World War II delayed research for close to a decade. Results of early experiments indicated that a cancer-selective carrier would need to be developed and it was not until the 1970s that monoclonal antibodies became available for this purpose. Unlike iodine, astatine shows a tendency to dehalogenate from molecular carriers such as these, particularly at sp3 carbon sites (less so from sp2 sites). Given the toxicity of astatine accumulated and retained in the body, this emphasized the need to ensure it remained attached to its host molecule. While astatine carriers that are slowly metabolized can be assessed for their efficacy, more rapidly metabolized carriers remain a significant obstacle to the evaluation of astatine in nuclear medicine. Mitigating the effects of astatine-induced radiolysis of labeling chemistry and carrier molecules is another area requiring further development. A practical application for astatine as a cancer treatment would potentially be suitable for a "staggering" number of patients; production of astatine in the quantities that would be required remains an issue.
Animal studies show that astatine, similarly to iodine, although to a lesser extent, is preferentially concentrated in the thyroid gland. Unlike iodine, astatine also shows a tendency to be taken up by the lungs and spleen, possibly because of in-body oxidation of At– to At+. If administered in the form of a radiocolloid it tends to concentrate in the liver. Experiments in rats and monkeys suggest that astatine-211 causes much greater damage to the thyroid gland than does iodine-131, with repetitive injection of the nuclide resulting in necrosis and cell dysplasia within the gland. Early research suggested that injection of astatine into female rodents caused morphological changes in breast tissue; this conclusion remained controversial for many years. General agreement was later reached that this was likely caused by the effect of breast tissue irradiation combined with hormonal changes due to irradiation of the ovaries.