
 | ||
Sequence homology is the biological homology between protein or DNA sequences, defined in terms of shared ancestry in the evolutionary history of life. Two segments of DNA can have shared ancestry either because of a speciation event (orthologs), or because of a duplication event (paralogs).
Contents
- Orthology
- Databases of orthologous genes
- Paralogy
- Paralogous regions
- Ohnology
- Xenology
- Gametology
- References
Homology among proteins or DNA is typically inferred from their sequence similarity. Significant similarity is strong evidence that two sequences are related by divergent evolution of a common ancestor. Alignments of multiple sequences are used to indicate which regions of each sequence are homologous.
The term "percent homology" is often used to mean "sequence similarity". The percentage of identical residues (percent identity) or the percentage of residues conserved with similar physicochemical properties (percent similarity), e.g. leucine and isoleucine, is usually used to "quantify the homology". Based on the definition of homology specified above this terminology is incorrect since sequence similarity is the observation, homology is the conclusion. Sequences are either homologous or not. As with anatomical structures, high sequence similarity might occur because of convergent evolution, or, as with shorter sequences, by chance, meaning that they are not homologous. Homologous sequence regions are also called conserved. This is not to be confused with conservation in amino acid sequences, where the amino acid at a specific position has been substituted with a different one that has functionally equivalent physicochemical properties.
Partial homology can occur where a segment of the compared sequences has a shared origin, while the rest does not. Such partial homology may result from a gene fusion event.
Orthology
Homologous sequences are orthologous if they are inferred to be descended from the same ancestral sequence separated by a speciation event: when a species diverges into two separate species, the copies of a single gene in the two resulting species are said to be orthologous. Orthologs, or orthologous genes, are genes in different species that originated by vertical descent from a single gene of the last common ancestor. The term "ortholog" was coined in 1970 by the molecular evolutionist Walter Fitch.
For instance, the plant Flu regulatory protein is present both in Arabidopsis (multicellular higher plant) and Chlamydomonas (single cell green algae). The Chlamydomonas version is more complex: it crosses the membrane twice rather than once, contains additional domains and undergoes alternative splicing. However it can fully substitute the much simpler Arabidopsis protein, if transferred from algae to plant genome by means of genetic engineering. Significant sequence similarity and shared functional domains indicate that these two genes are orthologous genes, inherited from the shared ancestor.
Orthology is strictly defined in terms of ancestry. Given that the exact ancestry of genes in different organisms is difficult to ascertain due to gene duplication and genome rearrangement events, the strongest evidence that two similar genes are orthologous is usually found by carrying out phylogenetic analysis of the gene lineage. Orthologs often, but not always, have the same function.
Orthologous sequences provide useful information in taxonomic classification and phylogenetic studies of organisms. The pattern of genetic divergence can be used to trace the relatedness of organisms. Two organisms that are very closely related are likely to display very similar DNA sequences between two orthologs. Conversely, an organism that is further removed evolutionarily from another organism is likely to display a greater divergence in the sequence of the orthologs being studied.
Databases of orthologous genes
Given their tremendous importance for biology and bioinformatics, orthologous genes have been organized in several specialized databases that provide tools to identify and analyze orthologous gene sequences. These resources employ approaches that can be generally classified into those that use heuristic analysis of all pairwise sequence comparisons, and those that use phylogenetic methods. Sequence comparison methods were first pioneered in the COGs database in 1997. These methods have been extended and automated in the following databases:
Tree-based phylogenetic approaches aim to distinguish speciation from gene duplication events by comparing gene trees with species trees, as implemented in databases such as
A third category of hybrid approaches uses both heuristic and phylogenetic methods to construct clusters and determine trees, for example
Paralogy
Homologous sequences are paralogous if they were created by a duplication event within the genome. For gene duplication events, if a gene in an organism is duplicated to occupy two different positions in the same genome, then the two copies are paralogous.
Paralogous genes often belong to the same species, but this is not necessary: for example, the hemoglobin gene of humans and the myoglobin gene of chimpanzees are paralogs. Paralogs can be split into in-paralogs (paralogous pairs that arose after a speciation event) and out-paralogs (paralogous pairs that arose before a speciation event). Between-species out-paralogs are pairs of paralogs that exist between two organisms due to duplication before speciation, whereas within-species out-paralogs are pairs of paralogs that exist in the same organism, but whose duplication event happened before speciation. Paralogs typically have the same or similar function, but sometimes do not: due to lack of the original selective pressure upon one copy of the duplicated gene, this copy is free to mutate and acquire new functions.
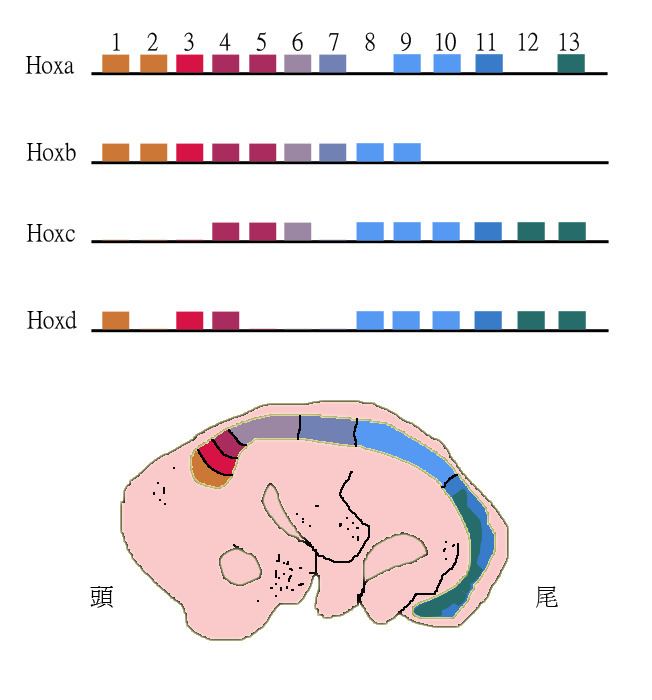
Paralogous genes can shape the structure of whole genomes and thus explain genome evolution to a large extent. Examples include the Homeobox (Hox) genes in animals. These genes not only underwent gene duplications within chromosomes but also whole genome duplications. As a result Hox genes in most vertebrates are clustered across multiple chromosomes with the HoxA-D clusters being the best studied.
Another example are the globin genes which encode myoglobin and hemoglobin are considered to be ancient paralogs. Similarly, the four known classes of hemoglobins (hemoglobin A, hemoglobin A2, hemoglobin B, and hemoglobin F) are paralogs of each other. While each of these proteins serves the same basic function of oxygen transport, they have already diverged slightly in function: fetal hemoglobin (hemoglobin F) has a higher affinity for oxygen than adult hemoglobin. Function is not always conserved, however. Human angiogenin diverged from ribonuclease, for example, and while the two paralogs remain similar in tertiary structure, their functions within the cell are now quite different.
It is often asserted that orthologs are more functionally similar than paralogs of similar divergence, but several papers have challenged this notion.
Paralogous regions
Sometimes, large chromosomal regions share gene content similar to other chromosomal regions within the same genome. They are well characterised in the human genome, where they have been used as evidence to support the 2R hypothesis. Sets of duplicated, triplicated and quadruplicated genes, with the related genes on different chromosomes, are deduced to be remnants from genome or chromosomal duplications. A set of paralogy regions is together called a paralogon. Well-studied sets of paralogy regions include regions of human chromosome 2, 7, 12 and 17 containing Hox gene clusters, collagen genes, keratin genes and other duplicated genes, regions of human chromosomes 4, 5, 8 and 10 containing neuropeptide receptor genes, NK class homeobox genes and many more gene families, and parts of human chromosomes 13, 4, 5 and X containing the ParaHox genes and their neighbors. The Major histocompatibility complex (MHC) on human chromosome 6 has paralogy regions on chromosomes 1, 9 and 19. Much of the human genome seems to be assignable to paralogy regions.
Ohnology
Ohnologous genes are paralogous genes that have originated by a process of whole-genome duplication. The name was first given in honour of Susumu Ohno by Ken Wolfe. Ohnologues are useful for evolutionary analysis because all ohnologues in a genome have been diverging for the same length of time (since their common origin in the whole genome duplication).
Xenology
Homologs resulting from horizontal gene transfer between two organisms are termed xenologs. Xenologs can have different functions, if the new environment is vastly different for the horizontally moving gene. In general, though, xenologs typically have similar function in both organisms. The term was coined by Walter Fitch.
Gametology
Gametology denotes the relationship between homologous genes on non-recombining, opposite sex chromosomes. Gametologs result from the origination of genetic sex determination and barriers to recombination between sex chromosomes. Examples of gametologs include CHDW and CHDZ in birds.