
 | ||
A primer is a short strand of RNA or DNA (generally about 18-22 bases) that serves as a starting point for DNA synthesis. It is required for DNA replication because the enzymes that catalyze this process, DNA polymerases, can only add new nucleotides to an existing strand of DNA. The polymerase starts replication at the 3'-end of the primer, and copies the opposite strand.
Contents
- Mechanism in vivo
- Primer removal
- Uses of synthetic primers
- PCR primer design
- Degenerate primers
- References
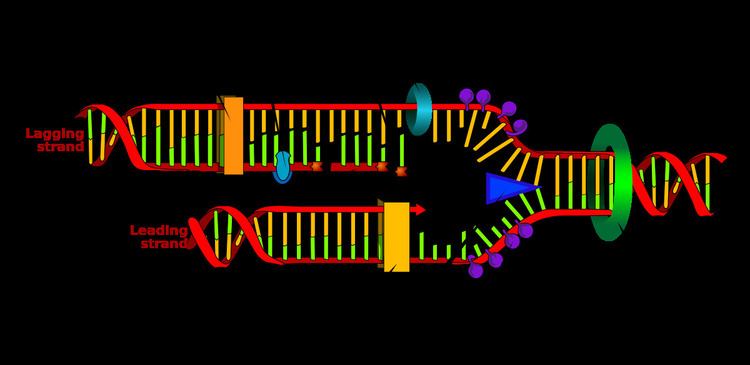
In vivo DNA replication utilizes short strands of RNA called RNA primers to initiate DNA synthesis on both the leading and lagging strands — DNA primers are not seen in vivo in humans. These RNA primers can be made de novo.
On the other hand, many of the in vitro laboratory techniques that involve DNA polymerase in biochemistry and molecular biology (such as DNA sequencing and the polymerase chain reaction), use DNA primers because they are more temperature stable. In experiments, it is often important to use a primer with a similar Tm (melting temperature) to the template strand it will be hybridizing to. A primer with a Tm significantly higher than the reaction's annealing temperature may mishybridize and extend at an incorrect location along the DNA sequence, while one with a Tm significantly lower than the annealing temperature may fail to anneal and extend at all. These primers are usually short, chemically synthesized oligonucleotides, with a length of about twenty bases. They are hybridized to a target DNA, which is then copied by the polymerase.
Mechanism in vivo
The lagging strand of DNA is that strand of the DNA double helix that is orientated in a 5' to 3' manner. Therefore, its complement must be synthesized in a 3'→5' manner. Because DNA polymerase III cannot synthesize in the 3'→5' direction, the lagging strand is synthesized in short segments known as Okazaki fragments. Along the lagging strand's template, primase builds RNA primers in short bursts. DNA polymerases are then able to use the free 3'-OH groups on the RNA primers to synthesize DNA in the 5'→3' direction.
The RNA fragments are then removed by DNA polymerase I for prokaryotes or DNA polymerase δ for eukaryotes (different mechanisms are used in eukaryotes and prokaryotes) and new deoxyribonucleotides are added to fill the gaps where the RNA was present. DNA ligase then joins the deoxyribonucleotides together, completing the synthesis of the lagging strand.
Primer removal
In eukaryotic primer removal, DNA polymerase δ extends the Okazaki fragment in 5' to 3' direction, and when it encounters the RNA primer from the previous Okazaki fragment, it displaces the 5′ end of the primer into a single-stranded RNA flap, which is removed by nuclease cleavage. Cleavage of the RNA flaps involves either flap structure-specific endonuclease 1 (FEN1) cleavage of short flaps, or coating of long flaps by the single-stranded DNA binding protein replication protein A (RPA) and sequential cleavage by Dna2 nuclease and FEN1.
This mechanism is a potential explanation of how the HIV virus can transform its genome into double-stranded DNA from the RNA-DNA formed after reverse transcription of its RNA. However, the HIV-encoded reverse transcriptase has its own ribonuclease activity that degrades the viral RNA during the synthesis of cDNA, as well as DNA-dependent DNA polymerase activity that copies the sense cDNA strand into antisense DNA to form a double-stranded DNA intermediate.
Uses of synthetic primers
DNA sequencing is used to determine the nucleotides in a DNA strand. The Sanger chain termination method of sequencing uses a primer to start the chain reaction.
In PCR, primers are used to determine the DNA fragment to be amplified by the PCR process. The length of primers is usually not more than 30 (usually 18–24) nucleotides, and they need to match the beginning and the end of the DNA fragment to be amplified. They direct replication towards each other – the extension of one primer by polymerase then becomes the template for the other, leading to an exponential increase in the target segment.
It is worth noting that primers are not always for DNA synthesis, but can in fact be used by viral polymerases, e.g. influenza, for RNA synthesis.
PCR primer design
Pairs of primers should have similar melting temperatures since annealing in a PCR occurs for both simultaneously. A primer with a Tm (melting temperature) significantly higher than the reaction's annealing temperature may mishybridize and extend at an incorrect location along the DNA sequence, while Tm significantly lower than the annealing temperature may fail to anneal and extend at all.
Primer sequences need to be chosen to uniquely select for a region of DNA, avoiding the possibility of mishybridization to a similar sequence nearby. A commonly used method is BLAST search whereby all the possible regions to which a primer may bind can be seen. Both the nucleotide sequence as well as the primer itself can be BLAST searched. The free NCBI tool Primer-BLAST integrates primer design and BLAST search into one application, as do commercial software products such as ePrime and Beacon Designer. Computer simulations of theoretical PCR results (Electronic PCR) may be performed to assist in primer design.
Many online tools are freely available for primer design, some of which focus on specific applications of PCR. The popular tools Primer3Plus and PrimerQuest can be used to find primers matching a wide variety of specifications. Highly degenerate primers for targeting a wide variety of DNA templates can be interactively designed using GeneFISHER. Primers with high specificity for a subset of DNA templates in the presence of many similar variants can be designed using DECIPHER.
Mononucleotide and dinucleotide repeats should be avoided, as loop formation can occur and contribute to mishybridization. Primers should not easily anneal with other primers in the mixture (either other copies of same or the reverse direction primer); this phenomenon can lead to the production of 'primer dimer' products contaminating the mixture. Primers should also not anneal strongly to themselves, as internal hairpins and loops could hinder the annealing with the template DNA.
When designing a primer for use in TA cloning, efficiency can be increased by adding AG tails to the 5' and the 3' end.
The reverse primer has to be the reverse complement of the given cDNA sequence. The reverse complement can be easily determined, e.g. with online calculators.
Degenerate primers
Sometimes degenerate primers are used. These are actually mixtures of similar, but not identical primers. They may be convenient if the same gene is to be amplified from different organisms, as the genes themselves are probably similar but not identical. The other use for degenerate primers is when primer design is based on protein sequence. As several different codons can code for one amino acid, it is often difficult to deduce which codon is used in a particular case. Therefore, primer sequence corresponding to the amino acid isoleucine might be "ATH", where A stands for adenine, T for thymine, and H for adenine, thymine, or cytosine, according to the genetic code for each codon, using the IUPAC symbols for degenerate bases. Use of degenerate primers can greatly reduce the specificity of the PCR amplification. The problem can be partly solved by using touchdown PCR.
Degenerate primers are widely used and extremely useful in the field of microbial ecology. They allow for the amplification of genes from thus far uncultivated microorganisms or allow the recovery of genes from organisms where genomic information is not available. Usually, degenerate primers are designed by aligning gene sequencing found in GenBank. Differences among sequences are accounted for by using IUPAC degeneracies for individual bases. PCR primers are then synthesized as a mixture of primers corresponding to all permutations.
There are a number of programs available to perform these primer predictions;