
ICD-9-CM 571.6 DiseasesDB 10615 | ICD-10 K74.3 OMIM 109720 | |
 | ||
Synonyms Primary biliary cirrhosis | ||
Primary biliary cholangitis (PBC), also known as primary biliary cirrhosis, is an autoimmune disease of the liver. It results from a slow, progressive destruction of the small bile ducts of the liver, causing bile and other toxins to build up in the liver, a condition called cholestasis. Further slow damage to the liver tissue can lead to scarring, fibrosis, and eventually cirrhosis.
Contents
- Signs and symptoms
- Causes
- Diagnosis
- Liver biopsy
- Histopathology stages
- Treatment
- Epidemiology
- Prognosis
- History
- PBC Foundation
- PBCers Organization
- Name
- References
Common symptoms are tiredness, itching and, in more advanced cases, jaundice. In early cases, there may only be changes in blood tests.
PBC is a relatively rare disease, affecting up to 1 in 3–4,000 people. It is much more common in women, with a sex ratio of at least 9:1 female to male.
The condition has been recognised since at least 1851 and was named "primary biliary cirrhosis" in 1949. Because cirrhosis is a feature only of advanced disease, a change of its name to "primary biliary cholangitis" was proposed by patient advocacy groups in 2014.
Signs and symptoms
People with PBC experience fatigue (80%) that leads to sleepiness during the daytime; more than half of those have severe fatigue. Itching (pruritus) occurs in 20–70%. People with more severe PBC may have jaundice (yellowing of the eyes and skin). PBC impairs bone density and there is an increased risk of fracture. Xanthelasma (skin lesions around the eyes) or other xanthoma may be present as a result of increased cholesterol levels.
PBC can eventually progress to cirrhosis of the liver. This in turn may lead to a number of symptoms or complications:
People with PBC may also sometimes have the findings of an associated extrahepatic autoimmune disorder such as rheumatoid arthritis or Sjögren's syndrome (in up to 80% of cases).
Causes
The cause of the disease is attributed to an immunological basis for the disease, making it an autoimmune disorder. It results from a slow, progressive destruction of the small bile ducts of the liver, with the intralobular ducts and the Canals of Hering (intrahepatic ductules) being affected early in the disease. This progresses to the development of fibrosis, cholestasis and, in some people, cirrhosis.
Most people with PBC (>90%) have anti-mitochondrial antibodies (AMAs) against pyruvate dehydrogenase complex (PDC-E2), an enzyme complex that is found in the mitochondria. People who are 'negative' for AMAs are usually found to be positive when more sensitive methods of detection are used.
Many PBC patients have a concomitant autoimmune disease, including rheumatological, endocrinological, gastrointestinal, pulmonary, or dermatological conditions, which suggests shared genetic and immune abnormalities. Common associations include Sjögren's syndrome, systemic sclerosis, rheumatoid arthritis, lupus, hypothyroidism and gluten sensitive enteropathy.
A genetic predisposition to disease has been thought to be important for some time. Evidence for this includes cases of PBC in family members, identical twins both having the condition (concordance), and clustering of PBC with other autoimmune diseases. In 2009, a Canadian-led group of investigators reported in the New England Journal of Medicine results from the first PBC genome-wide association study. This research revealed parts of the IL12 signaling cascade, particularly IL12A and IL12RB2 polymorphisms, to be important in the aetiology of the disease in addition to the HLA region. In 2012, two independent PBC association studies increased the total number of genomic regions associated to 26, implicating many genes involved in cytokine regulation such as TYK2, SH2B3 and TNFSF11.
An environmental Gram negative alphabacterium — Novosphingobium aromaticivorans has been associated with this disease with several reports suggesting an aetiological role for this organism. The mechanism appears to be a cross reaction between the proteins of the bacterium and the mitochondrial proteins of the liver cells. The gene encoding CD101 may also play a role in host susceptibility to this disease.
There is a failure of immune tolerance against the mitochondrial pyruvate dehydrogenase complex (PDC-E2), and this may also be the case with other proteins including the gp210 and p62 nuclear pore proteins. Gp210 has increased expression in the bile duct of anti-gp210 positive patients, and these proteins may be associated with prognosis.
Diagnosis
To diagnose PBC, it needs to be distinguished from other conditions with similar symptoms, such as autoimmune hepatitis or primary sclerosing cholangitis (PSC).
Most patients can be diagnosed without invasive investigation, as the combination of anti-mitochondrial antibodies and typical (cholestatic) liver enzyme tests are considered diagnostic. However, a liver biopsy is needed to determine the stage of disease.
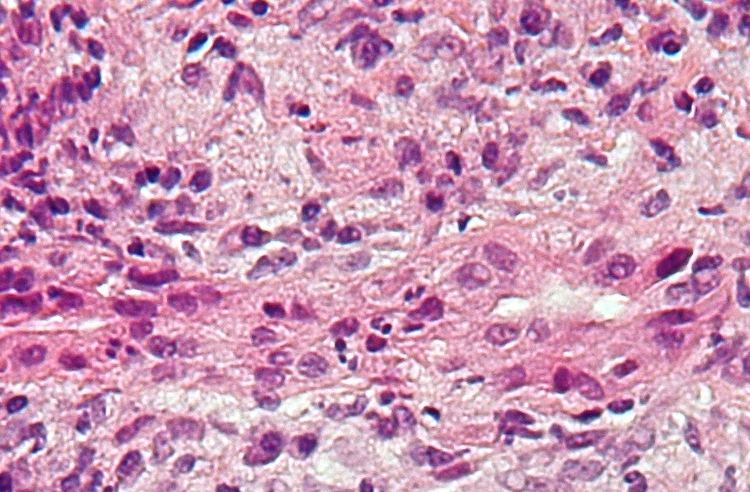
Liver biopsy
On microscopic examination of liver biopsy specimens, PBC is characterized by interlobular bile duct destruction. These histopathologic findings in primary biliary cholangitis include the following:
Histopathology stages
Treatment
There is no known cure, but medication may slow the progression so that a normal lifespan and quality of life may be attainable for many patients.
As in all liver diseases, consumption of alcohol is contraindicated.
In advanced cases, a liver transplant, if successful, results in a favorable prognosis.
The farnesoid X receptor agonist, obeticholic acid, which is a modified bile acid, was approved by the United States Food and Drug Administration on May 27, 2016, as an orphan drug in an accelerated approval program, based on its reduction in the level of the biomarker alkaline phosphatase, as a surrogate endpoint for clinical benefit. It is indicated for the treatment of primary biliary cholangitis in combination with ursodeoxycholic acid in adults with an inadequate response to UDCA, or as monotherapy in adults unable to tolerate UDCA. Additional studies are being required to prove its clinical benefit.
Epidemiology
PBC is a chronic autoimmune liver disease with a female gender predominance with female:male ratio is at least 9:1 and a peak incidence in the fifth decade of life. In some areas of the US and UK, the prevalence is estimated to be as high as 1 in 4000. This is much more common than in South America or Africa, which may be due to better recognition in the US and UK. First-degree relatives may have as much as a 500 times increase in prevalence, but there is debate if this risk is greater in the same generation relatives or the one that follows.
Prognosis
The serum bilirubin level is an indicator of the prognosis of PBC, with levels of 2–6 mg/dL having a mean survival time of 4.1 years, 6–10 mg/dL having 2.1 years and those above 10 mg/dL having a mean survival time of 1.4 years.
After liver transplant, the recurrence rate may be as high as 18% at 5 years, and up to 30% at 10 years. There is no consensus on risk factors for recurrence of the disease.
Complications of PBC can be related to chronic cholestasis or cirrhosis of the liver. Chronic cholestasis leads to osteopenic bone disease and osteoporosis, alongside hyperlipidaemia and vitamin deficiencies.
Patients with PBC have an increased risk of hepatocellular carcinoma compared to the general population, as is found in other cirrhotic patients. In patients with advanced disease, one series found an incidence of 20% in men and 4% in women.
History
In 1851, Addison and Gull described the clinical picture of progressive obstructive jaundice in the absence of mechanical obstruction of the large bile ducts. Although most sources credit Ahrens with coining the term in 1950, Dauphinee and Sinclair had used the name primary biliary cirrhosis for this disease in 1949. The association with anti-mitochondrial antibodies was first reported in 1965 and their presence was recognized as a marker of early, pre-cirrhotic disease.
PBC Foundation
The PBC Foundation is a UK-based international charity offering support and information to people with PBC, their families and friends. It campaigns for increasing recognition of the disorder, improved diagnosis and treatments, and estimates over 8000 people are undiagnosed in the UK. The Foundation has supported research into PBC including the development of the PBC-40 quality of life measure published in 2004 and helped establish the PBC Genetics Study. It was founded by Collette Thain in 1996, after she was diagnosed with the condition. Thain was awarded an MBE Order of the British Empire in 2004 for her work with the Foundation. The PBC Foundation helped initiate the name change campaign in 2014.
PBCers Organization
The PBCers Organization is a US-based non-profit patient support group that was founded in 1996 and advocates for greater awareness of the disease and new treatments. It has supported the initiative for a change in name.
Name
In 2014 the PBC Foundation, with the support of the PBCers Organization, the PBC Society (Canada) and other patient groups, advocated a change in name from "primary biliary cirrhosis" to "primary biliary cholangitis", citing that most PBC patients did not have cirrhosis and that this often had negative connotations of alcoholism. Patient and professional groups were canvassed. Support for this name change came from professional bodies including the American Association for the Study of Liver Diseases and the European Association for the Study of the Liver. Advocates for the name change published calls to adopt the new name in multiple hepatology journals in the fall of 2015.