
Entrez 3145 | Ensembl ENSG00000256269 | |
 | ||
External IDs OMIM: 609806 MGI: 96112 HomoloGene: 158 GeneCards: HMBS | ||
Porphobilinogen deaminase (hydroxymethylbilane synthase, or uroporphyrinogen I synthase) is an enzyme (EC 2.5.1.61) that in humans is encoded by the HMBS gene. Porphobilinogen deaminase is involved in the third step of the heme biosynthetic pathway. It catalyzes the head to tail condensation of four porphobilinogen molecules into the linear hydroxymethylbilane while releasing four ammonia molecules:
Contents
4 porphobilinogen + H2OStructure and function
Functionally, porphobilinogen deaminase catalyzes the loss of ammonia from the porphobilinogen monomer (deamination) and its subsequent polymerization to a linear tetrapyrrole, which is released as hydroxymethylbilane:
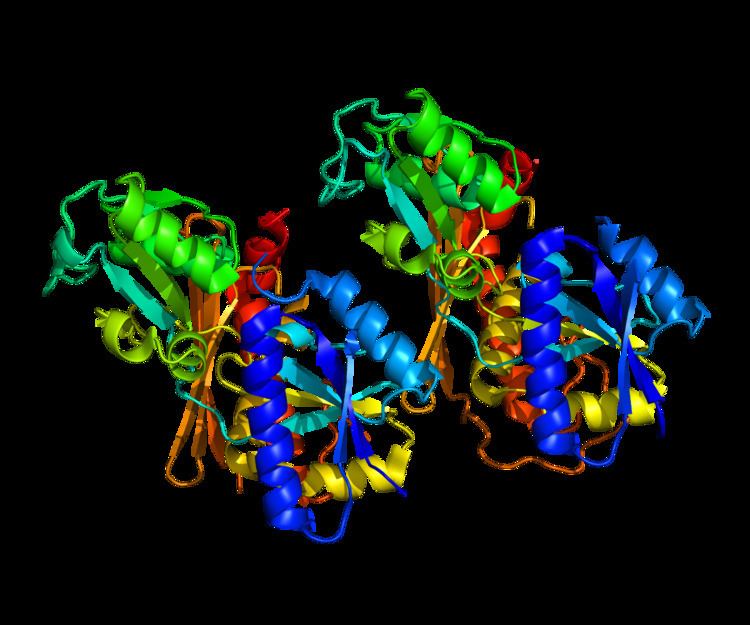
The structure of 40-42 kDa porphobilinogen deaminase, which is highly conserved amongst organisms, consists of three domains. Domains 1 and 2 are structurally very similar: each consisting of five beta-sheets and three alpha helices in humans. Domain 3 is positioned between the other two and has a flattened beta-sheet geometry. A dipyrrole, a cofactor of this enzyme consisting of two condensed porphobilinogen molecules, is covalently attached to domain 3 and extends into the active site, the cleft between domains 1 and 2. Several positively charged arginine residues, positioned to face the active site from domains 1 and 2, have been shown to stabilize the carboxylate functionalities on the incoming porphobilinogen as well as the growing pyrrole chain. These structural features presumably favor the formation of the final hydroxymethylbilane product. Porphobilinogen deaminase usually exists in dimer units in the cytoplasm of the cell.
Reaction mechanism
The first step is believed to involve an E1 elimination of ammonia from porphobilinogen, generating a carbocation intermediate (1). This intermediate is then attacked by the dipyrrole cofactor of porphobilinogen deaminase, which after losing a proton yields a trimer covalently bound to the enzyme (2). This intermediate is then open to further reaction with porphobilinogen (1 and 2 repeated three more times). Once a hexamer is formed, hydrolysis allows hydroxymethylbilane to be released, as well as cofactor regeneration (3).
Pathology
The most well-known health issue involving porphobilinogen deaminase is acute intermittent porphyria, an autosomal dominant genetic disorder where insufficient hydroxymethylbilane is produced, leading to a build-up of porphobilinogen in the cytoplasm. This is caused by a gene mutation that, in 90% of cases, causes decreased amounts of enzyme. However, mutations where less-active enzymes and/or different isoforms have been described.