
 | ||
Perineuronal nets (PNNs) are specialized extracellular matrix structures responsible for synaptic stabilization in the adult brain. PNNs are found around certain neuron cell bodies and proximal neurites in the central nervous system. PNNs play a critical role in the closure of the childhood critical period, and their digestion can cause restored critical period-like synaptic plasticity in the adult brain. They are largely negatively charged and composed of chondroitin sulfate proteoglycans, molecules that play a key role in development and plasticity during postnatal development and in the adult.
Contents
- History
- Composition
- Neuroprotection
- Restriction of AMPA receptor mobility
- Buffering system for cations
- Role in neuroplasticity
- Ocular dominance plasticity
- Fear memories
- Developmental song learning
- Epilepsy
- Stroke
- Alzheimers disease
- References
PNNs appear to be mainly present in the cortex, hippocampus, thalamus, brainstem, and the spinal cord. Studies of the rat brain have shown that the cortex contains high numbers of PNNs in the motor and primary sensory areas and relatively fewer in the association and limbic cortices. In the cortex, PNNs are associated mostly with inhibitory interneurons and are thought to be responsible for maintaining the excitatory/inhibitory balance in the adult brain.
History
The existence of PNNs has been inferred by Golgi, Lugaro, Donaggio, Martinotti, Ramón y Cajal and Meyer. However, Ramón y Cajal credits Golgi with the discovery of PNNs because he was the first to draw attention to them and gave the first precise description in 1893. Moreover, Golgi brought interest to the subject due to his opinion that the PNN was not a neuronal structure but rather a "kind of corset of neurokeratin which impeded the spread of current from cell to cell". Despite debating the topic, Ramón y Cajal claimed that the perineuronal net was simply a staining artifact derived from the coagulation of extracellular fluids. Due to his influential opinion at the time, interest in the topic subsided.
Interest arose in the 1960s when several authors drew attention to the presence of periodic-acid-Schiff-positive (PAS-positive) material surrounding nerve cells. This PAS-positive material was suspected of being composed of negatively charged substances, such as chondroitin sulfate proteoglycans (CSPGs). However, the authors clung to the idea that the material was intricately connected to the blood–brain barrier and failed to see the similarities it had with the perineuronal net described by Golgi. Interest only rose again in the past few decades when it was discovered that PNNs constitute markers for physiologically mature neurons.
Composition
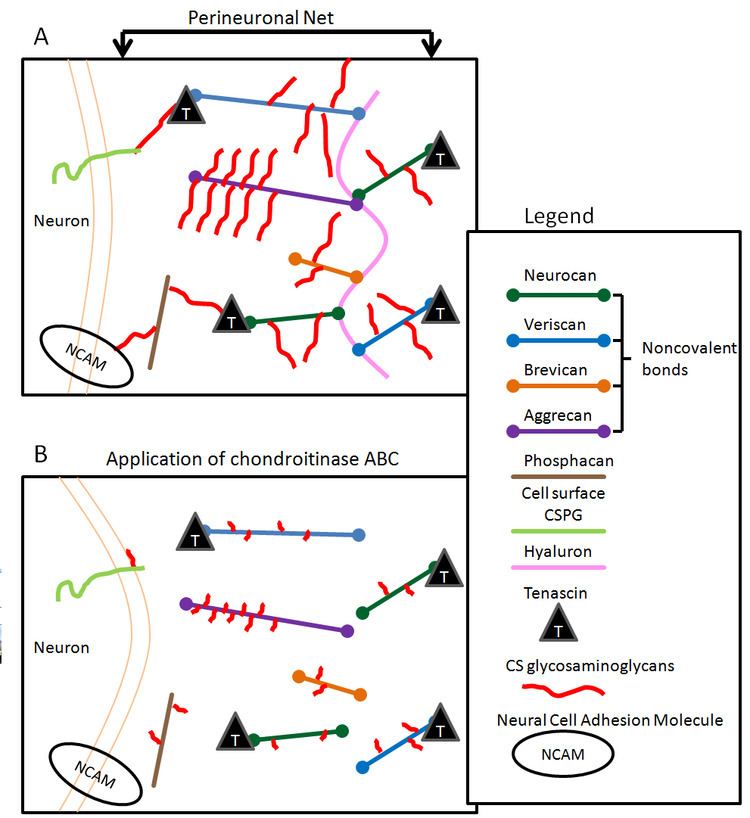
PNNs are composed of a condensed matrix of chondroitin sulfate proteoglycans, molecules that consist of a core protein and a glycosaminoglycan (GAG) chain. The CS-GAG chains associated with PNNs differs from those found floating in the extra-cellular matrix in a noncondensed form. PNNs are composed of brevican, neurocan, versican, aggrecan, phosphacan, hyaluronan, tenascin-R and various link proteins. The CSPGs aggrecan, versican, neurocan, brevican, and phosphacan are bound to hyaluronan. PNNs found in both the brain and the spinal cord have the same composition. Chondroitinase ABC (ChABC), a bacterial enzyme routinely used to digest CSPGs, works by catalyzing the removal of the CS-GAG chains of CSPGs.
In the cortex and other subcortical areas, PNNs preferentially surround GABAergic interneurons containing the calcium-binding protein parvalbumin. The onset of the critical period corresponds closely to the emergence of parvalbumin-positive cells. Parvalbumin-positive cells synapse onto α1-subunit-containing GABAA receptors. The α1-subunit-containing GABAA receptors have been shown to be the only GABAA receptors that drive cortical plasticity. For this reason, PNNs were first thought to have a strong role in the closure of the critical period.
Neuroprotection
A fine regulation of axonal and dendritic growth is required in the adult CNS to preserve important connections while still allowing for structural plasticity. This function has been recognized to be mediated by several myelin-associated proteins and CSPGs. In order to assess the physiological role of PNNs in the undamaged CNS, ChABC was injected in the healthy cerebellum of adult rats. In the site of ChABC injections, there was profuse outgrowth of the terminal branches of Purkinje cell neurons. However, myelinated axon segments were not affected and remained normal. Purkinje axon sprouting was first evident four days after the degradation of CSPGs. Within 42 days, the expression of CSPGs gradually recovered, at which point axon outgrowth regressed, indicating that there was no significant formation of stable synaptic contacts. While CSPGs are very important in neuroprotection, this indicates that CSPGs may not be the only molecules important for the preservation of anatomical plasticity.
Restriction of AMPA receptor mobility
Cell surface proteins, including neurotransmitter receptors, are highly mobile in the plasma membrane due to lateral diffusion. Fast movements of AMPA-type glutamate receptors (AMPARs) are involved in the modulation of synaptic transmission. As a receptor is used, it becomes desensitized and unable to operate efficiently for a short period of time. Diffusion of the desensitized receptor for the exchange of a naive functional one increases synaptic fidelity during fast repetitive stimulation. PNNs compartmentalize the neuronal surface and act as lateral diffusion barriers for AMPARs, limiting synaptic exchange. This may be part of the reason that synaptic plasticity is limited once PNNs become upregulated.
Buffering system for cations
Most of the parvalbumin-positive neurons surrounded by PNNs also contain the potassium channel Kv3.1b subunit. These specific cells have been identified as fast-spiking cells. These neurons have a low input resistance of the cell membrane, a high resting membrane potential, a short duration of both action potentials and the refractory period, a high firing frequency, and an almost constant amplitude of their action potentials. It appears that both Kv3.1 channels and PNNs are both required for the fast-spiking behavior of these neurons. These potassium channels are important because outward potassium currents are responsible for the repolarization of the cell membrane during an action potential. It has been shown that Kv3.1 currents allow a neuron to follow a high frequency stimulation and/or to generate high firing rates without spike adaption, characteristics that fit well with fast-spiking cells. This characteristic of the cells is important as blockade of the Kv3.1b channel has been shown to slow the rate of ocular dominance plasticity.
PNNs, with their strongly negative charge, may serve as cation exchangers preventing the free diffusion of potassium or sodium ions. Due to the spatial, temporal, and numerical disproportions between Na+ influx and K+ efflux, the PNN provides a possible buffering system for extracellular cations. However, this hypothesis has yet to be proven.
Role in neuroplasticity
PNNs play an important role in neuroplasticity. Traumatic injury of the CNS results in degeneration of denervated and damaged neurons, the formation of a glial scar, and collateral sprouting of surviving neurons. PNNs have been shown to be inhibitory to axonal regeneration and outgrowth. CSPGs are the main axon growth inhibitory molecules in the glial scar that play a role in the failure of the axon to regenerate after injury. In the rat brain and spinal cord, the expression of various CSPGs (brevican, versican, neurocan, and NG2) increases after injury. In vivo treatment with ChABC results in the enhancement of the regeneration of axons (specifically dopaminergic neurons) and the promotion of axon regeneration and functional recovery following spinal cord injury.
CSPGs and PNNs are also implicated in the restricted plasticity present after CNS injury. In the rat cerebellum, application of ChABC promotes structural plasticity of Purkinje axons. Following spinal cord injury, rats treated with ChABC show structural and functional recovery in the form of increased regrowth of axons into the denervated territory and the recovery of motor and bladder function. Plasticity of intact areas in the brain stem and spinal cord also increases following spinal cord injury.
Ocular dominance plasticity
The critical period is a stage when a necessary amount of experience is required for the proper organization of a neural pathway. The absence of this experience may lead to the permanent formation of incorrect connections. The classic model of the critical period has been the visual system. Normally, the primary visual cortex contains neurons organized in ocular dominance columns, with groups of neurons responding preferentially to one eye or the other. If an animal's dominant eye is sutured early in life and kept sutured through the visual critical period (monocular deprivation), the cortex permanently responds preferentially to the eye that was kept open, resulting in ocular dominance shift. However, if the eye is sutured after the critical period, the shift does not occur.
In rats, digestion of PNNs using the bacterial enzyme chondroitinase ABC reactivates the visual critical period. Specifically, digestion of PNNs in the visual cortex well after the closure of the critical period (postnatal day 70) reactivated critical period plasticity and allowed ocular dominance shift to occur. However, the effects of monocular deprivation in the reactivated case were not as strong as monocular deprivation during a normal critical period. Additionally, in adult rats that had been monocularly deprivated since youth, digestion of PNNs brought about a full structural and functional recovery (recovery of ocular dominance, visual acuity, and dendritic spine density). However, this recovery only occurred once the open eye was sutured to allow the cortical representation of the deprived eye to recover.
Fear memories
Fear conditioning in animals is used to model anxiety disorders such as PTSD. Fear conditioning works by pairing an initially neutral stimulus with an aversive stimulus, leading to long-lasting fear memories. In an adult animal, fear conditioning induces a permanent memory resilient to erasure by extinction. After extinction, conditioned fear responses can recover spontaneously after a reexposure to the aversive stimulus. In contrast, in early postnatal development, extinction of a conditioned fear response leads to memory erasure. The organization of PNNs in the amygdala coincides with this switch in fear memory resilience. In the adult animal, degradation of PNNs in the amygdala with ChABC renders subsequently acquired fear memories susceptible to erasure. Extinction training was necessary for the loss of fear behavior. Additionally, fear memories acquired before the degradation of the PNNs were not affected by their degradation.
Developmental song learning
Developmental song learning is a model used for the sensorimotor critical period. Birdsong learning in the zebra finch occurs during a critical period similar to that for human speech. This critical period occurs in two parts. The first consists of an early perceptual phase in which sounds are merely memorized. This is followed by a second sensorimotor phase in which feedback is used to shape proper sounds. In the song nuclei HVC, over 80% of PNNs surround parvalbumin-positive neurons. The presence of perineuronal nets predicts the maturity of a zebra finch's song, with greater PNN density indicating a more mature song and likely greater synaptic stability. Unlike the visual critical period, extensive preliminary investigation has shown that degrading the PNNs with ChABC does not reopen the critical period of sensorimotor plasticity. This can be attributed to the additional complicating factors present in a sensorimotor system compared to a purely sensory system. In humans, complications in the speech sensorimotor critical period is implicated in disorders such as autism. Reopening of the critical period in zebra finches may lead to discoveries leading to treatment for these disorders.
Epilepsy
Epilepsy is a chronic neurological disorder characterized by abnormal electrical activity in the brain. This abnormal electrical activity results in increased plastic changes that play a part of the pathogenesis of the disease. Following seizures, there is a decrease in phosphacan and phosphacan-positive PNNs and an increase in cleaved brevican in the temporal lobe and hippocampus. Seizures also increase the amount of full-length neurocan, a CSPG only found in the neonatal brain. This degradation of CSPGs and PNNs could be responsible for the increased plasticity associated with the disorder.
Stroke
Following stroke, there is some increased plasticity resulting in the restoration of some function. In the rat model, following a cortical lesion, there is a reduction of PNNs in the region surrounding the infarction. Specifically, there is a reduction in the CSPGs aggrecan, versican, and phosphacan and an accumulation of full-length neurocan. This downregulation of PNNs also occurs in brain regions as distant as the thalamus. The degradation of PNNs may be responsible for the increased plasticity seen post-stroke. One issue with typical stroke recovery is the typical period of increased plasticity is generally not long enough to allow stroke patients acceptable recovery of function. One possible treatment strategy may be to degrade PNNs for a longer period of time to allow for greater recovery.
Alzheimer's disease
There appear to be several roles for CSPGs in Alzheimer's disease. PNNs may provide protection against excitotoxicity, oxidative stress, and the formation of neurofibrillary tangles. There have been conflicting reports as to the number of PNNs in the human Alzheimer's brain, with some studies reporting a reduction and others reporting no change. There is no clear consensus as the susceptibility of parvalbumin-positive neurons, the majority of neurons surrounded by PNNs. However, PNNs have been found to localize with both amyloid plaques and neurofibrillary tangles. Since amyloid plaques have been implicated in the progression of Alzheimer's disease, this suggests that PNNs are either instrumental in their formation or are a reaction to their formation. In vitro studies have shown that CSPGs promote beta amyloid fibril formation. Since beta amyloid is a strong stimulant to CSPG production and CSPGs are inhibitory to neuronal growth and synaptic plasticity, this may lead to the decreased axon density and synaptic loss in Alzheimer's disease.