
Specialty oncology ICD-O M9540/0-9550 MeSH D009455 | ICD-10 D36.1 (ILDS D36.160) DiseasesDB 23371 | |
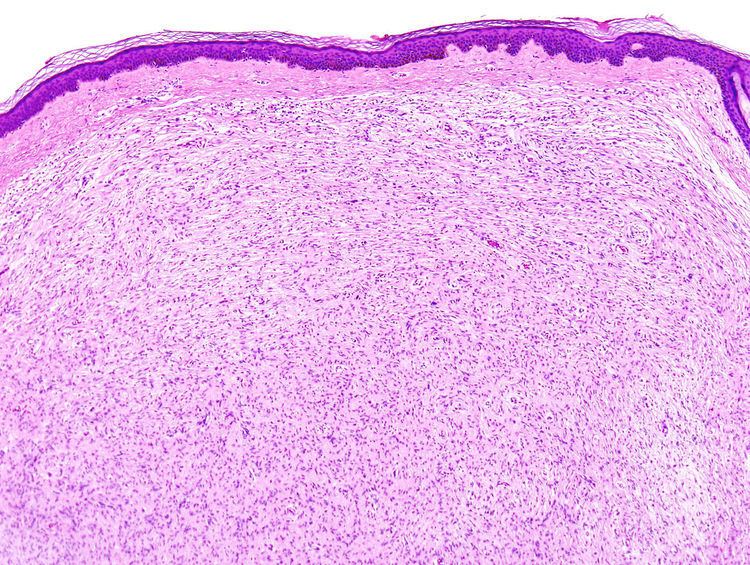 | ||
A neurofibroma is a benign nerve sheath tumor in the peripheral nervous system. In 90% of cases they're found as stand-alone tumors, while the remainder are found in persons with neurofibromatosis type I (NF1), an autosomal dominant genetically inherited disease, they can result in a range of symptoms from physical disfiguration and pain to cognitive disability. Neurofibromas arise from nonmyelinating-type Schwann cells that exhibit biallelic inactivation of the NF1 gene that codes for the protein neurofibromin. This protein is responsible for regulating the RAS-mediated cell growth signaling pathway. In contrast to schwannomas, another type of tumor arising from Schwann cells, neurofibromas incorporate many additional types of cells and structural elements in addition to Schwann cells, making it difficult to identify and understand all the mechanisms through which they originate and develop.
Contents
Types
Neurofibromas have been subdivided into two broad categories: dermal and plexiform. Dermal neurofibromas are associated with a single peripheral nerve, while plexiform neurofibromas are associated with multiple nerve bundles. According to the World Health Organization classification system, dermal and plexiform neurofibromas are grade I tumors. Plexiform neurofibroma are more difficult to treat and can transform into malignant tumors. Dermal neurofibroma do not become malignant.
Anatomy
Dermal neurofibromas (sometimes referred to as cutaneous neurofibromas) originate in nerves in the skin. Three kinds are distinguished:
Age of onset
Dermal neurofibroma typically arise in the teenage years and are often associated with the onset of puberty. They continue to increase in number and size throughout adulthood, although there are limits to how big they get.
Medical complications
Dermal neurofibromas can lead to stinging, itching, pain and disfiguration.
There is no evidence of malignant transformation.
Anatomy
Plexiform neurofibromas can grow from nerves in the skin or from more internal nerve bundles, and can be very large. Internal plexiform neurofibromas are very difficult to remove completely because they extend through multiple layers of tissue and the attempt would damage healthy tissue or organs.
Age of onset
Plexiform neurofibromas occur earlier in life and are thought to be congenital defects.
Medical complications
Plexiform neurofibroma can cause disfigurement, neurological and other clinical deficits.
Plexiform neurofibromas have the potential to cause severe clinical complications if they occur in certain areas.
About 10% of plexiform neurofibromas undergo transformation into a malignant peripheral nerve sheath tumor (MPNST). The formation of malignant cancers from neurofibromas is associated with the loss of expression of the CDKN2A or TP53 gene in non-myelinating Schwann cells that also exhibit biallelic inactivation of the NF1 gene.
Cause
This section discusses the tumorigenesis of neurofibroma in terms of genetics, cell signaling, histology and the cell cycle.
Neurofibromin 1 gene
The NF1 gene is composed of 60 exons spanning 350kb of genomic data, and maps to chromosomal region 17qll.2. This gene codes for neurofibromin which is a large 220-250 KDa cytoplasmic protein that is composed of 2,818 amino acids with three alternatively spliced exons (9a, 23a and 48a) in the encoding gene. The functional part of neurofibromin is a GAP, or GTPase-activating protein. GAP accelerates the conversion of the active GTP-bound RAS to its inactive GDP-bound form, inactivating RAS and reducing RAS-mediated growth signaling. Loss of RAS control leads to increased activity of other signaling pathways including RAF, ERK1/2, PI3K, PAK and mTOR-S6 kinase. It is suspected that this increased activity of downstream RAS pathways might work together to increase cell growth and survival. Genes that code for proteins that regulate cell growth, such as NF1 and TP53, are referred to as tumor suppressor genes. Neurofibromin has other growth-regulatory properties besides its ability to regulate RAS activity, but these other functions are poorly understood at this time.
Schwann cells
There are two kinds of Schwann cells, myelinating and nonmyelinating. While myelinating Schwann cells cover large diameter (>1 micrometer) peripheral nervous system (PNS) axons with myelin, nonmyelinating Schwann cells encapsulate small diameter PNS axons with their cytoplasmic processes. Nonmyelinating Schwann cells are the neoplastic element in neurofibromas. This conglomeration of nonmyelinating Schwann cells and axons is called a Remak bundle.
While nonmyelinating Schwann cells are the origin of neurofibromas, the mutations that make them susceptible to this transformation occur in Schwann cell precursors during early nerve development. Mutated nonmyelinating Schwann cells do not form normal Remak bundles. Instead, they fail to properly surround and segregate target axons. It is unknown at this time why, if both types of Schwann cells exhibit bilallelic inactivation of the NF1 gene, only the nonmyelinating variety give rise to neurofibromas.
Loss of tumor suppressor function
Neurofibromas arise from nonmyelinating Schwann cells that only express the inactive version of the NF1 gene, which leads to a complete loss of expression of functional neurofibromin. While one defective allele may be inherited, loss of heterozygosity (LOH) must occur before a neurofibroma can form; this is called the ‘two-hit hypothesis’. This LOH happens by the same mechanisms, such as oxidative DNA damage, that causes mutations in other cells.
Once a nonmyelinating Schwann cell has suffered inactivation of its NF1 genes, it begins to proliferate rapidly. This condition is called hyperplasia, which is cell growth beyond what is normally seen. However, despite increased numbers of nonmyelinating Schwann cells, there is no neurofibroma yet. In order for the neurofibroma to develop, cells that are heterozygous for the NF1 gene must be recruited to the site. It has been hypothesized that the proliferating nonmyelinating Schwann cells secrete chemoattractants such as the KIT ligand, and angiogenic factors such as the heparin-binding growth factor midkine. These chemicals promote the migration of different kinds of cells that are heterozygous for the NF1 gene into the hyperplastic lesions created by the nonmyelinating Schwann cells. These cell types include fibroblasts, perineurial cells, endothelial cells, and mast cells. The mast cells then secrete mitogens or survival factors that alter the developing tumor microenvironment and result in neurofibroma formation.
Dermal and plexiform neurofibromas differ in later development stages, but the details are unclear at this point.
Diagnosis
A blood test for protein melanoma inhibitory activity may be used to detect the presence of neurofibromas.
Dermal neurofibroma
Dermal neurofibromas are not usually surgically removed unless they are painful or disfiguring, because there are generally so many of them and they are not dangerous.
CO2 lasers have been used to remove dermal neurofibromas. In a paper titled Hypertrophic Scars After Therapy with CO2 Laser for Treatment of Multiple Cutaneous Neurofibromas Ostertag et al. said this about treatment by laser: “The cosmetic disfigurement is the most important issue in the decision to treat cutaneous symptoms of neurofibromatosis. Treating patients with extensive neurofibromas with [a] CO2 laser is still the best choice. However, it is strongly advised that a test treatment be performed to judge the effectiveness of the procedure and whether the developed scar is an acceptable trade-off.”
Surgery
As of 2002, the primary treatment option for plexiform neurofibroma was surgery.
Removal of plexiform neurofibromas is difficult because they can be large and cross tissue boundaries. However, besides pain, plexiform neurofibromas are sometimes removed due to the possibility of malignant transformation.
The following examples show that plexiform neurofibromas can form anywhere and can make surgical resection difficult:
Radiation
Once a plexiform neurofibroma has undergone malignant transformation, radiation and chemotherapy can be used as treatment. However, radiation is generally not used as a treatment for plexiform neurofibromas because of concerns that this could actually promote malignant transformation. There has even been a documented case of a Schwannoma being induced from a neurofibroma due to radiation therapy.
Medications
ACE inhibitors have been proposed as a novel treatment of neurofibromas. ACE inhibitors are currently used to treat hypertension and congestive heart failure, to avert remodeling and reinfarction after myocardial infarction, and to ameliorate diabetic nephropathy and other renal diseases. ACE inhibitors work by indirectly down regulating TGF-beta, which is a growth factor that has been shown to influence the development of tumors.
No effect
Pirfenidone inhibits fibroblast growth. Studies showed no improvement over controls.
Tipifarnib (also known as drug R115777) inhibits the activation of RAS. This drug is a Farnesyltransferase inhibitor which inhibits the Ras kinase in a post translational modification step before the kinase pathway becomes hyperactive. It successfully passed phase one clinical trials but was suspended (NCT00029354) in phase two after showing no improvement over controls.
Research
There are many drug therapies under study for neurofibromas. These are in various stages of research; more time will be required to determine if these are viable options for the treatment of neurofibromas.
The combination of erlotinib with sirolimus is being studied to treat low-grade gliomas.
Early research has shown potential for using the c-kit tyrosine kinase blocking properties of imatinib to treat plexiform neurofibromas.
Peginterferon alfa-2b is being studied to treat plexiform neurofibromas.
Sirolimus is an antibiotic developed as an antifungal agent. It inhibits mTOR signalling. It is being studied to treat plexiform neurofibromas.
Sorafenib is being studied for treatment of unresectable plexiform neurofibroma and low-grade astrocytomas.
In vitro, tranilast, inhibits growth of neurofibroma cells.
Gene therapy for the neurofibromin 1 gene represents the ultimate solution to preventing the cluster of maladies which are enabled by the mutation. As of 2006, therapy for NF1 tumors had not been tested due to the lack of an appropriate NF1 tumor model.