
 | ||
In chemistry, metal-catalysed hydroboration is a reaction used in organic synthesis. It is one of several examples of homogeneous catalysis.
Contents
History
In 1975, Kono and Ito reported that Wilkinson's catalyst (Rh(PPh3)3Cl) can undergo oxidative addition with catecholborane (HBcat) or 4,4,6-trimethyl-1,3,2-dioxaborinane. These two borane compounds are otherwise slow to participate in hydroboration. In 1985, Männig and Nöth demonstrated for the first time that Wilkinson’s catalyst indeed catalyzes hydroboration of alkenes with HBcat.
Whereas uncatalyzed hydroboration using HBcat leads to reduction of the carbonyl group, the catalyzed version is selective for the alkene.
As indicated by subsequent research, transition metal-catalyzed hydroboration proceeds with attractive functional group-, regio-, stereo-, and chemo- selectivity.
Mechanism
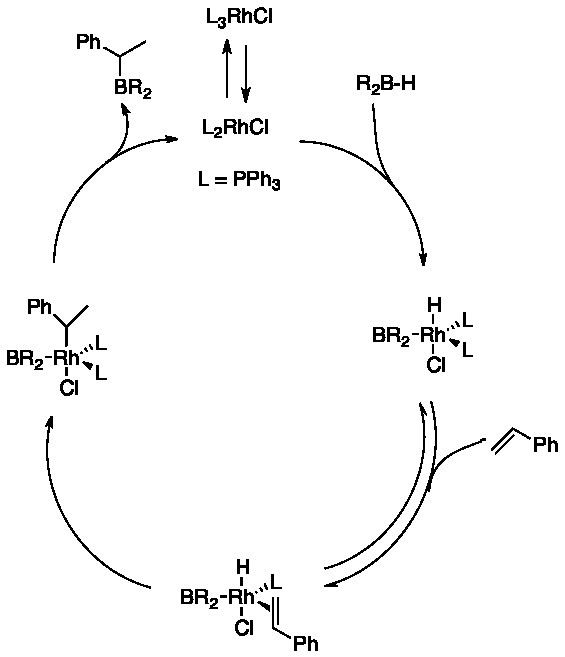
The rhodium-catalyzed hydroboration reaction is thought to be initiated with the dissociation of a triphenylphosphine from the Rh(I) centre. Oxidative addition of the B-H bond of the borane reagent to this 14 e− species is then followed by coordination of the alkene to the 16e− Rh(III) hydride complex. Subsequent migratory insertion of the alkene into the rhodium-hydride bond can give two regioisomeric alkyl rhodium(III) boride complexes. Reductive elimination of the boronate ester regenerates the catalyst. Catalyst prepared and handled under anaerobic condition reverses the selectivity to favor the secondary boronate ester. What has been debated is the coordination of the alkene. In the dissociative mechanism, proposed by Männig and Nöth, and supported by Evans and Fu the coordination is accompanied by the loss of one triphenylphosphine ligand.
In the associative mechanism (see below), proposed by Burgess et al., the alkene binds trans to the chloride without dissociation of a triphenylphosphine ligand. The mechanism has been studied by computational methods. Dorigo and Schleyer excluded the associative mechanism by an ab initio study on the dissociative mechanism, whereas Musaev and co-workers support the associative mechanism.
Selectivity
Apart from the original evidence provided by Männig and Nöth, the total synthesis of (+)-ptilocaulin also demonstrates selective hydroboration of a terminal alkene in the presence of a ketone.
In terms of regioselectivity, the catalyzed hydroboration differs from the uncatalyzed parallel. Depending on the ligands and the alkene, either Markovnikov or anti-Markovnikov product result. The difference in regioselectivity is more pronounced in the hydroboration of vinylarenes with HBcat. Wilkinson’s catalyst or the cation Rh(COD)2 (in the presence of PPh3) produces the Markovnikov product. The anti-Markovnikov product is produced in the absence of a catalyst. It is worth noticing that the use of RhCl3·nH2O produces selectively the anti-Markovnikov product. To account for the high regioselectivity of catalyzed hydroboration, Hayashi proposed a mechanism involving a η3-benzylrhodium complex.
Catalyzed hydroboration-oxidation of substituted alkenes can be rendered enantioselective. In 1990, Brown and co-workers achieved asymmetric hydroboration using an achiral catalyst and chiral borane sources derived from ephedrine and pseudoephedrine. In most cases, the regioselectivity was poor although the ee values can be close to 90%.
Use of a chiral catalyst and an achiral borane source is more common, e.g. chiral diphosphines such as BINAP.
Styrene or its simple derivatives are usually the prochiral substrate.
Enantioselectivity tends to be lowered with ortho-substituents on the aromatic ring, as well as further substitution on the olefin. Successful results have also been obtained on other reactants. The second class of ligands is phosphinamine ligands. In 1993, Brown first reported the successful use of QUINAP in asymmetric alkene hydroboration. QUINAP improve upon the intolerance of substitution on the aromatic ring as observed for diphosphine ligands. Reactions using styrene and derivatives with electron-donating groups on the para position still gave high ee values. Similar results were also obtained on cyclic vinyl arenes. Such results expand the scope of asymmetric hydroboration to more sterically demanding alkenes. Several new ligands of this class have also been developed. Some recent results are summarized below.
The studies above have all utilized oxidation of the boronate esters to produce alcohols, which is a severe limitation to the synthetic scope of such species, especially when they can be made enantioselectively. Another important class of compounds that can be derived from boronate esters is α-substituted benzylamines, some of which are commercially useful. The synthesis of such chiral amines via catalytic hydroboration involves conversion of the catecholboronate ester to trialkylborane by diethyl zinc or methylmagnesium chloride. Reaction of the trialkylborane with hydroxylamine-O-sulfonic acid produces primary benzylamines. Secondary amines can also be prepared by in situ formation of N-chloramines.