
Entrez 51205 | Ensembl ENSG00000162836 | |
 | ||
External IDs MGI: 1931010 HomoloGene: 41128 GeneCards: ACP6 | ||
Lysophosphatidic acid phosphatase type 6 is an acid phosphatase enzyme that is encoded in humans by the ACP6 gene.
Contents
- Structure
- Mechanism
- Function
- Disease relevance
- Ovarian cancer
- Gauchers Disease
- Related gene defects
- Interactions
- References
It acts as a phosphomonoesterase at low pHs. It is responsible for the hydrolysis of Lysophosphatidic acids (LPAs) to their respective monoacylglycerols and the release a free phosphate group in the process. The enzyme has higher activity for myristate-LPA (14 carbon chain), oleate-LPA (18 carbon chain and one unsaturated carbon-carbon bond), laurate-LPA (12 carbon chain) or palmitate-LPA (16 carbon chain). When the substrate is stearate-LPA (18 carbon chain), the enzyme has reduced activity. Phosphatidic acids can also be hydrolyzed by lysophosphatidic acid phosphatase, but at a significantly lower rate. The addition of the second fatty chain makes fitting into the active site much harder.
LPAs are necessary for healthy cell growth, survival and pro-angiogenic factors for both in vivo and in vitro cells. Unbalanced concentrations of lysophosphatidic acid phosphatase can frequently lead to unbalanced LPA concentrations, which can cause metabolic disorders, and lead to ovarian cancer in women.
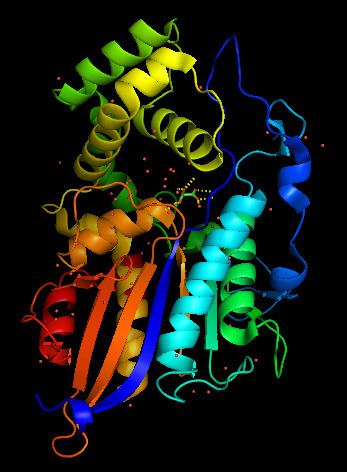
Structure
Lysophosphatidic acid phosphatase is a monomer composed of two domains. One domain functions as a cap on the enzyme, while the second comprises the body of the enzyme. The enzyme has two (α) alpha helices on one side, seven (β) beta sheets in the middle, and two more α helices on the opposite side. The space between the two domains serves as a large substrate pocket, as well as a channel through which water molecules can move through. This channel is lined with hydrophilic residues that lead the water molecule to the active site, where the terminal water molecule interacts with Asp-335 residue and is then activated. This catalyzes the bond formation to the phosphate group. Lysophosphatidic acid phosphatase also has two disulfide bridges. One that binds α12 and α4 together, and the other that binds a turn at the edge of β7 strand. Analysis of the pocket shows that the active site pocket has space for one long fatty acid chain, but not for two fatty chains, furthermore supporting that this enzyme has strong preference for LPAs.
The active site of lysophosphatidic acid phosphatase has six main residues required to stabilize the phosphate group and the hydroxyl. These residues are Arg-58, His-59, Arg-62, Arg-168, His-334, Asp-335. Though there are no crystal structures with a LPA molecule in the substrate pocket, the crystal structure with malonate shows the hydrogen bonding between the enzyme residues and the carbonyl groups that would stabilize the phosphate and hydroxyl groups on the LPA. In the active site, the phosphate group is stabilized by Arg-58, Arg-62, Arg-168 and His-334. The guanidinium groups and hydrogen on the protonated imidazole ring from the histidine residue. When any of these residues were mutated to alanine, the catalytic activity of the enzyme was greatly reduced. This is evidence that the active site requires this "claw" to hold on to the phosphate group, the aspartic acid residue to activate a water molecule, and the histidine residue to provide a proton to form the alcohol. It should also be noted that when the residues at the entrance to the water channel were mutated to bulkier residues, such as Leucine, Phenylalanine or Tryptophan, the enzyme was no longer capable of hydrolyzing the LPA. This further supports the proposed mechanism in which water, supplied from the solvent through the channel, acts as a nucleophile in the active site.
Mechanism
Lysophosphatidic acid phosphatase has a very similar reaction mechanism to those of other phosphomonoesterases. One significant difference is this enzymes ability to perform the desired hydrolysis most effectively at low pHs. At low pHs, all the arganine and histidines are found in their protonated states. This ensures that Arg58, Arg62, Arg168 and His334 will be able to stabilize the phosphate group and hydroxyl group in the active site. The aspartic acid side chain has a pKa of approximately 4. In an acidic environment, this residue will readily give up its proton, but will also take a proton away from water if the side chain is deprotonized, thus catalyzing the hydroxyl attack on the phosphate group. Soon after the deprotonation of the histidine residue and the protonation of the aspartic acid residue, the histidine residue will deprotonate the aspartic acid residue, preparing the enzyme to hydrolyze an LPA again.
Function
Lysophosphatidic acid phophatase has several roles. Although lysophosphatidic acid phosphatase is found ubiquitously throughout the body with higher levels in the kidney, heart, small intestine, muscles and the liver, evidence suggests that this enzyme is regulates lipid metabolism in the mitochondria.
Another function is to control the concentration of LPAs that serve as messengers for G protein-coupled receptors in the cell. These LPAs are responsible for the signaling of cell growth, proliferation, muscle contractions, and wound healing, among many other roles. Due to this role, an imbalance in the concentrations of lysophosphatidic acid phosphatase can frequently lead to several metabolic diseases.
Lysophosphatidic acid phosphatase is also responsible for the digestion of lysophosphatidic acids when the cell enters a state of phosphate starvation. These enzymes break down LPAs and release phosphate groups. This stops the production of phospholipids and phosphatidic acids to signal the end of a cell's proliferation process.
Disease relevance
Two examples of disorders caused by irregular LPA levels and show increased enzyme activity are ovarian cancer and Gaucher's Disease.
Ovarian cancer
Lysophosphatidic acid phosphatase activity is used to detect and to quantify irregular levels of LPAs on a cell's surface. LPAs are receptor-active mediators that promote cell motility, cell growth and cell survival. There is clear evidence that cancerous ovarian cells have an increased level of LPA concentrations on their cell surfaces. These LPAs leak from the cell surface into the blood stream. The high levels of LPAs in the blood are used as tumor markers. In these cell clusters, lysophosphatidic acid phosphatase activity is higher than it is in regular cells. This can be attributed to the significantly increased levels of LPA that are secreted and synthesized by the ovarian cancer cells. This helps explain the cancerous cell's radical behavior and uncontrollable proliferation caused by the imbalance of enzyme and substrate concentrations, therefore leading to the inability to turn off the LPA cascade signalling effectively. One possible way to address and treat ovarian cancer cell proliferation would be to increase the concentration of lysophosphatidic acid phosphatase on the cell's surface, thus decreasing the amount of LPAs available to signal the cell to proceed with its radical behavior.
Gaucher's Disease
Gaucher's Disease is another disorder in which lysophosphatidic acid phosphatase is found in irregular concentrations. Increased concentration levels of lysophosphatidic acid phosphatase and enzyme activity in a patient's blood are used in order to aid in the diagnosis of Gaucher's Disease. The increased activity can be attributed to the excess of LPAs in the serum. Gaucher's Disease is caused by an accumulation of glucosphingolipids in the body tissues and bone marrow. LPAs are a precursor of sphingolipids, so although lysophosphatidic acid phosphatase is not directly responsible for the imbalance that leads to Gaucher's Disease, its activity can be used to support the diagnosis of the disease. It is important to note that even though the increased activity of the enzyme has been found in patients with Gaucher's Disease, there has been no clear relation between the enzyme and the progression of the disease.
Related gene defects
Interactions
ACP6 has been shown to interact with Integrin-linked kinase.