
Entrez 3643 | Ensembl ENSG00000171105 | |
 | ||
External IDs OMIM: 147670 MGI: 96575 HomoloGene: 20090 GeneCards: INSR | ||
The insulin receptor (IR) is a transmembrane receptor that is activated by insulin, IGF-I, IGF-II and belongs to the large class of tyrosine kinase receptors. Metabolically, the insulin receptor plays a key role in the regulation of glucose homeostasis, a functional process that under degenerate conditions may result in a range of clinical manifestations including diabetes and cancer. Biochemically, the insulin receptor is encoded by a single gene INSR, from which alternate splicing during transcription results in either IR-A or IR-B isoforms. Downstream post-translational events of either isoform result in the formation of a proteolytically cleaved α and β subunit, which upon combination are ultimately capable of homo or hetero-dimerisation to produce the ≈320 kDa disulfide-linked transmembrane insulin receptor.
Contents
Structure
Initially, transcription of alternative splice variants derived from the INSR gene are translated to form one of two monomeric isomers; IR-A in which exon 11 is excluded, and IR-B in which exon 11 is included. Inclusion of exon 11 results in the addition of 12 amino acids upstream of the intrinsic furin proteolytic cleavage site.
Upon receptor dimerisation, after proteolytic cleavage into the α- and β-chains, the additional 12 amino acids remain present at the C-terminus of the α-chain (designated αCT) where they are predicted to influence receptor–ligand interaction.
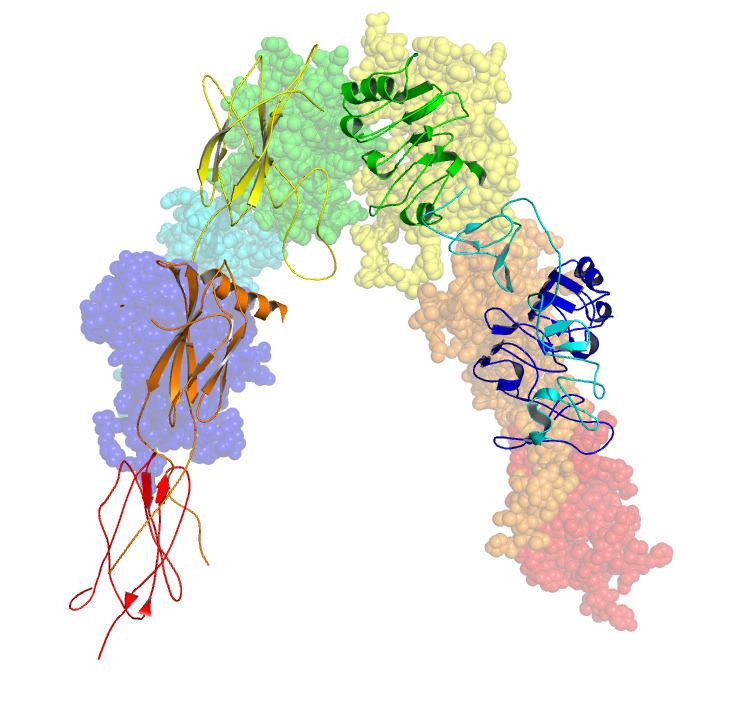
Each isometric monomer is structurally organized into 8 distinct domains consists of; a leucine-rich repeat domain (L1, residues 1-157), a cysteine-rich region (CR, residues 158-310), an additional leucine rich repeat domain (L2, residues 311-470), three fibronectin type III domains; FnIII-1 (residues 471-595), FnIII-2 (residues 596-808) and FnIII-3 (residues 809-906). Additionally, an insert domain (ID, residues 638-756) resides within FnIII-2, containing the α/β furin cleavage site, from which proteolysis results in both IDα and IDβ domains. Within the β-chain, downstream of the FnIII-3 domain lies a transmembrane helix (TH) and intracellular juxtamembrane (JM) region, just upstream of the intracellular tyrosine kinase (TK) catalytic domain, responsible for subsequent intracellular signaling pathways.
Upon cleavage of the monomer to its respective α- and β-chains, receptor hetero or homo-dimerisation is maintained covalently between chains by a single disulphide link and between monomers in the dimer by two disulphide links extending from each α-chain. The overall 3D ectodomain structure, possessing four ligand binding sites, resembles an inverted ‘V’, with the each monomer rotated approximately 2-fold about an axis running parallel to the inverted 'V' and L2 and FnIII-1 domains from each monomer forming the inverted 'V's apex.
Ligand binding
The insulin receptor's endogenous ligands include insulin, IGF-I and IGF-II. The binding of ligand to the α-chains of the IR ectodomain induces structural changes within the receptor leading to autophosphorylation of various tyrosine residues within the intracellular TK domain of the β-chain. These changes facilitate the recruitment of specific adapter proteins such as the insulin receptor substrate proteins (IRS) in addition to SH2-B (Src Homology 2 - B ), APS and protein phosphatases, such as PTP1B, eventually promoting downstream processes involving blood glucose homeostasis.
Strictly speaking the relationship between IR and ligand shows complex allosteric properties. This was indicated with the use of a Scatchard plots which identified that the measurement of the ratio of IR bound ligand to unbound ligand does not follow a linear relationship with respect to changes in the concentration of IR bound ligand, suggesting that the IR and its respective ligand share a relationship of cooperative binding. Furthermore, the observation that the rate of IR-ligand dissociation is accelerated upon addition of unbound ligand implies that the nature of this cooperation is negative; said differently, that the initial binding of ligand to the IR inhibits further binding to its second active site - exhibition of allosteric inhibition.
Although the precise binding mechanism of IR and its ligand has not yet been elucidated structurally, as identified using a systems biology approach, biologically relevant prediction of the IR-ligand kinetics (insulin/IGF-I) has been identified in the context of the currently available IR ectodomain structure.
These models state that each IR monomer possesses 2 insulin binding sites; site 1, which binds to the 'classical' binding surface of insulin: consisting of L1 plus αCT domains and site 2, consisting of loops at the junction of FnIII-1 and FnIII-2 predicted to bind to the 'novel' hexamer face binding site of insulin. As each monomer contributing to the IR ectodomain exhibits 3D 'mirrored' complementarity, N-terminal site 1 of one monomer ultimately faces C-terminal site 2 of the second monomer, where this is also true for each monomers mirrored complement (the opposite side of the ectodomain structure). Current literature distinguishes the complement binding sites by designating the second monomer's site 1 and site 2 nomenclature as either site 3 and site 4 or as site 1' and site 2' respectively. As such, these models state that each IR may bind to an insulin molecule (which has two binding surfaces) via 4 locations, being site 1, 2, (3/1') or (4/2'). As each site 1 proximally faces site 2, upon insulin binding to a specific site, 'crosslinking' via ligand between monomers is predicted to occur (i.e. as [monomer 1 Site 1 - Insulin - monomer 2 Site (4/2')] or as [monomer 1 Site 2 - Insulin - monomer 2 site (3/1')]). In accordance with current mathematical modelling of IR-insulin kinetics, there are two important consequences to the events of insulin crosslinking; 1. that by the aforementioned observation of negative cooperation between IR and its ligand that subsequent binding of ligand to the IR is reduced and 2. that the physical action of crosslinking brings the ectodomain into such a conformation that is required for intracellular tyrosine phosphorylation events to ensue (i.e. these events serve as the requirements for receptor activation and eventual maintenance of blood glucose homeostasis).
Biological significance
Tyrosine kinase receptors, including the insulin receptor, mediate their activity by causing the addition of a phosphate group to particular tyrosines on certain proteins within a cell. The "substrate" proteins that are phosphorylated by the Insulin Receptor include a protein called "IRS-1" for "insulin receptor substrate 1". IRS-1 binding and phosphorylation eventually leads to an increase in the high affinity glucose transporter (Glut4) molecules on the outer membrane of insulin-responsive tissues, including muscle cells and adipose tissue, and therefore to an increase in the uptake of glucose from blood into these tissues. In other words, the glucose transporter Glut4 is transported from cellular vesicles to the cell surface, where it then can mediate the transport of glucose into the cell.
Pathology
The main activity of activation of the insulin receptor is inducing glucose uptake. For this reason "insulin insensitivity", or a decrease in insulin receptor signaling, leads to diabetes mellitus type 2 – the cells are unable to take up glucose, and the result is hyperglycemia (an increase in circulating glucose), and all the sequelae that result from diabetes.
Patients with insulin resistance may display acanthosis nigricans.
A few patients with homozygous mutations in the INSR gene have been described, which causes Donohue syndrome or Leprechaunism. This autosomal recessive disorder results in a totally non-functional insulin receptor. These patients have low-set, often protuberant, ears, flared nostrils, thickened lips, and severe growth retardation. In most cases, the outlook for these patients is extremely poor, with death occurring within the first year of life. Other mutations of the same gene cause the less severe Rabson-Mendenhall syndrome, in which patients have characteristically abnormal teeth, hypertrophic gingiva (gums), and enlargement of the pineal gland. Both diseases present with fluctuations of the glucose level: After a meal the glucose is initially very high, and then falls rapidly to abnormally low levels.
Regulation of gene expression
The activated IRS-1 acts as a secondary messenger within the cell to stimulate the transcription of insulin-regulated genes. First, the protein Grb2 binds the P-Tyr residue of IRS-1 in its SH2 domain. Grb2 is then able to bind SOS, which in turn catalyzes the replacement of bound GDP with GTP on Ras, a G protein. This protein then begins a phosphorylation cascade, culminating in the activation of mitogen-activated protein kinase (MAPK), which enters the nucleus and phosphorylates various nuclear transcription factors (such as Elk1).
Stimulation of glycogen synthesis
Glycogen synthesis is also stimulated by the insulin receptor via IRS-1. In this case, it is the SH2 domain of PI-3 kinase (PI-3K) that binds the P-Tyr of IRS-1. Now activated, PI-3K can convert the membrane lipid phosphatidylinositol 4,5-bisphosphate (PIP2) to phosphatidylinositol 3,4,5-triphosphate (PIP3). This indirectly activates a protein kinase, PKB (Akt), via phosphorylation. PKB then phosphorylates several target proteins, including glycogen synthase kinase 3 (GSK-3). GSK-3 is responsible for phosphorylating (and thus deactivating) glycogen synthase. When GSK-3 is phosphorylated, it is deactivated, and prevented from deactivating glycogen synthase. In this roundabout manner, insulin increases glycogen synthesis.
Degradation of insulin
Once an insulin molecule has docked onto the receptor and effected its action, it may be released back into the extracellular environment or it may be degraded by the cell. Degradation normally involves endocytosis of the insulin-receptor complex followed by the action of insulin degrading enzyme. Most insulin molecules are degraded by liver cells. It has been estimated that a typical insulin molecule is finally degraded about 71 minutes after its initial release into circulation.
Interactions
Insulin receptor has been shown to interact with